Landelouci Karima, Sinha Shruti, Pépin Geneviève
Département de Biologie Médicale, Université du Québec à Trois-Rivières, Trois-Rivières, QC, Canada.
Groupe de Recherche en Signalisation Cellulaire, Université du Québec à Trois-Rivières, Trois-Rivières, QC, Canada.
Front Cell Infect Microbiol. 2022 Feb 7;12:820273. doi: 10.3389/fcimb.2022.820273. eCollection 2022.
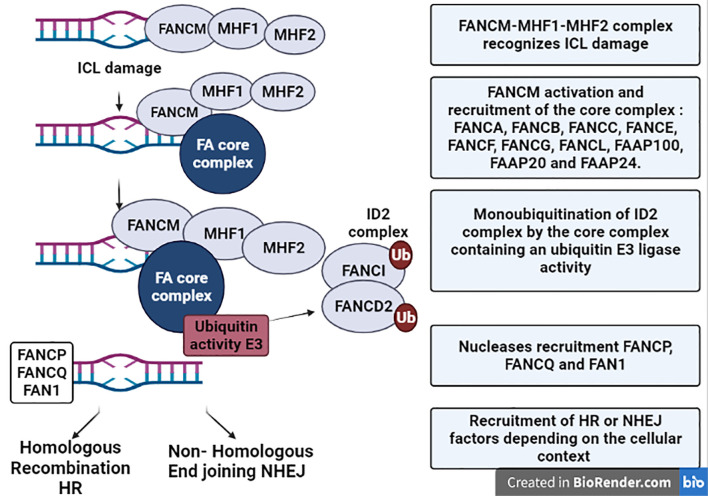
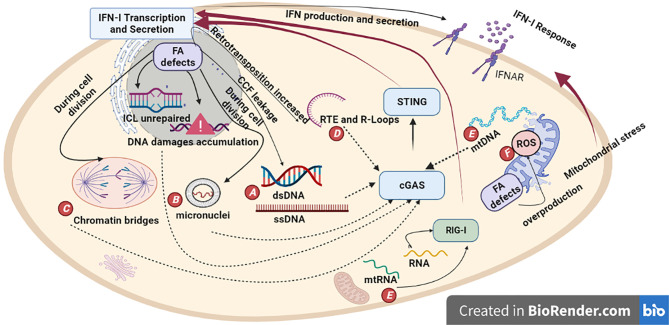
Fanconi Anemia (FA) is a genome instability syndrome caused by mutations in one of the 23 repair genes of the Fanconi pathway. This heterogenous disease is usually characterized by congenital abnormalities, premature ageing and bone marrow failure. FA patients also show a high predisposition to hematological and solid cancers. The Fanconi pathway ensures the repair of interstrand crosslinks (ICLs) DNA damage. Defect in one of its proteins prevents functional DNA repair, leading to the accumulation of DNA breaks and genome instability. Accumulating evidence has documented a close relationship between genome instability and inflammation, including the production of type-I Interferon. In this context, type-I Interferon is produced upon activation of pattern recognition receptors by nucleic acids including by the cyclic GMP-AMP synthase (cGAS) that detects DNA. In mouse models of diseases displaying genome instability, type-I Interferon response is responsible for an important part of the pathological symptoms, including premature aging, short stature, and neurodegeneration. This is illustrated in mouse models of Ataxia-telangiectasia and Aicardi-Goutières Syndrome in which genetic depletion of either Interferon Receptor IFNAR, cGAS or STING relieves pathological symptoms. FA is also a genetic instability syndrome with symptoms such as premature aging and predisposition to cancer. In this review we will focus on the different molecular mechanisms potentially leading to type-I Interferon activation. A better understanding of the molecular mechanisms engaging type-I Interferon signaling in FA may ultimately lead to the discovery of new therapeutic targets to rescue the pathological inflammation and premature aging associated with Fanconi Anemia.
范可尼贫血(FA)是一种基因组不稳定综合征,由范可尼通路的23个修复基因之一发生突变引起。这种异质性疾病通常表现为先天性异常、早衰和骨髓衰竭。FA患者还极易患血液系统和实体癌症。范可尼通路可确保修复链间交联(ICL)DNA损伤。其一种蛋白质的缺陷会阻止功能性DNA修复,导致DNA断裂积累和基因组不稳定。越来越多的证据表明基因组不稳定与炎症之间存在密切关系,包括I型干扰素的产生。在这种情况下,I型干扰素是在模式识别受体被包括检测DNA的环鸟苷酸-腺苷酸合成酶(cGAS)在内的核酸激活后产生的。在显示基因组不稳定的疾病小鼠模型中,I型干扰素反应是包括早衰、身材矮小和神经退行性变等重要病理症状的原因。共济失调毛细血管扩张症和艾卡迪-古铁雷斯综合征的小鼠模型说明了这一点,其中干扰素受体IFNAR、cGAS或STING的基因缺失可缓解病理症状。FA也是一种遗传不稳定综合征,具有早衰和易患癌症等症状。在本综述中,我们将重点关注可能导致I型干扰素激活的不同分子机制。更好地理解FA中参与I型干扰素信号传导的分子机制最终可能会发现新的治疗靶点,以挽救与范可尼贫血相关的病理性炎症和早衰。