Department of Rheumatology and Immunology, 159358Affiliated Hospital of Zunyi Medical University, Affiliated Hospital of Zunyi Medical University, Zunyi, Guizhou 563003, P. R. China.
School of Foreign Languages of Zunyi Medical University, Zunyi, Guizhou 563003, P. R. China.
J Int Med Res. 2022 Mar;50(3):3000605221084873. doi: 10.1177/03000605221084873.
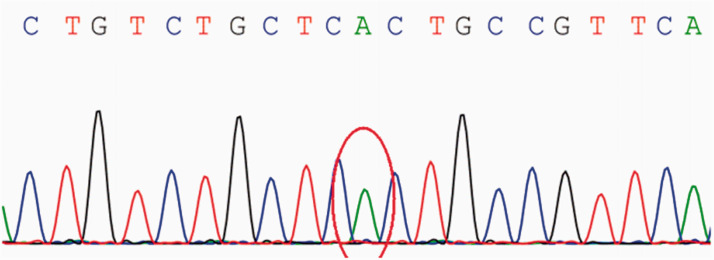
A 26-year-old Asian woman with persistent muscle weakness was diagnosed with polymyositis based on biopsy findings at another hospital 11 years ago. However, her symptoms fluctuated repeatedly under treatment with prednisone and immunosuppressive agents, and worsened 2 months prior to the current presentation. A second muscle biopsy suggested metabolic myopathy, and genetic testing revealed a novel c.1074C > T variant in the glycogen synthase 1 gene (), which is implicated in muscle glycogen storage disease type 0. However, no abnormalities in glycogen deposition were found by biopsy; rather, muscle fibers exhibited large intracellular lipid droplets. Furthermore, muscle strength was greatly restored and circulating levels of creatine kinase indicative of muscle degeneration greatly reduced by vitamin B2 treatment. Therefore, the final diagnosis was lipid storage myopathy.
一位 26 岁的亚裔女性,11 年前在另一家医院因肌肉无力进行了活检,被诊断为多发性肌炎。然而,在泼尼松和免疫抑制剂治疗下,她的症状反复波动,在本次就诊前 2 个月恶化。第二次肌肉活检提示代谢性肌病,基因检测显示糖原合酶 1 基因()的 c.1074C>T 新型变异,该变异与肌肉糖原贮积症 0 型相关。然而,活检并未发现糖原沉积异常;相反,肌纤维内有大量的细胞内脂质滴。此外,肌肉力量显著恢复,肌酸激酶循环水平显著降低,这表明肌肉退行性病变得到了极大的改善。因此,最终诊断为脂质贮积性肌病。