Paim-Marques Luciana, de Oliveira Rodrigo Joel, Appenzeller Simone
Department of Pediatrics, University of Florida, Gainesville, FL, USA.
Department of Orthopedics, Rheumatology and Traumatology- School of Medical Sciences and University of Campinas (UNICAMP), São Paulo, Brazil.
J Multidiscip Healthc. 2022 Mar 10;15:485-495. doi: 10.2147/JMDH.S290580. eCollection 2022.
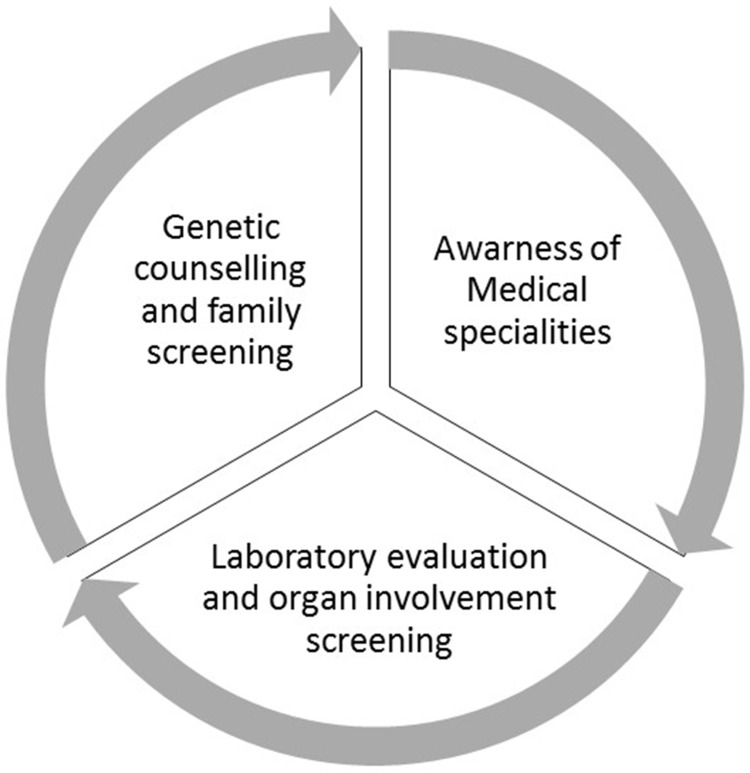
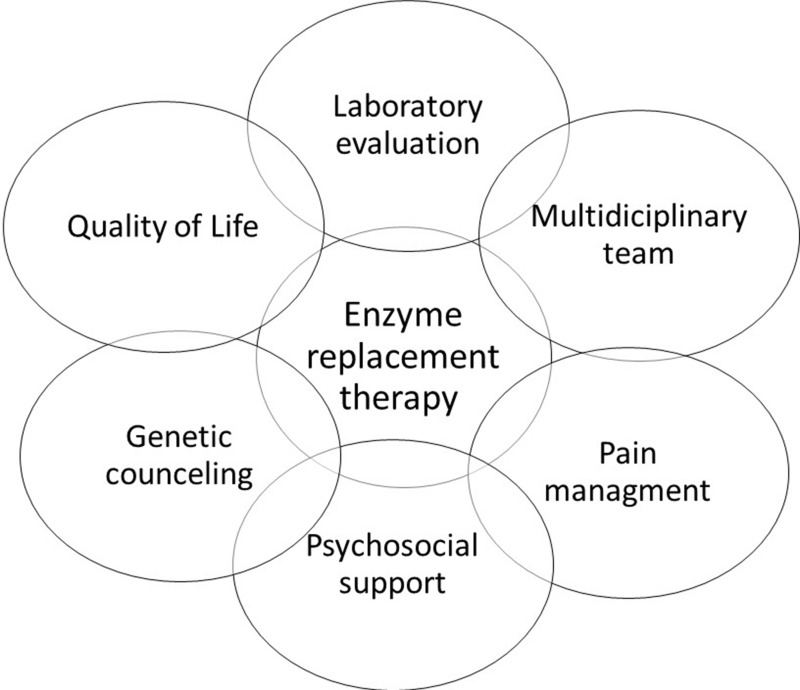
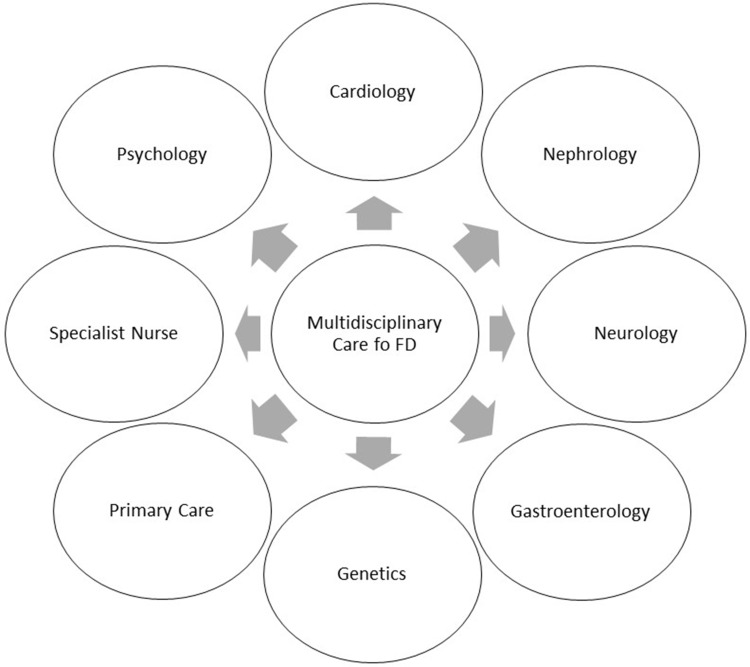
Fabry disease (FD) is a rare, recessive X-linked, multisystemic lysosomal storage disorder (LSD) that results from a deficiency in the hydrolase alpha-galactosidase A (α-GalA) caused by a gene variant. The progressive accumulation of the glycosphingolipid globotriaosylceramide (Gb3) in organs such as skin, kidney, brain, joints, vascular walls and eyes are responsible for the wide spectrum of clinical manifestations, often unspecific. In result, clinically relevant and life-threatening complications, such as malignant ventricular arrhythmia, sudden cardiac death, end stage kidney failure and stroke may occur. In this review, we will describe the clinical features and the current perspectives in the multidisciplinary management Of FD patients.
法布里病(FD)是一种罕见的、隐性X连锁多系统溶酶体贮积症(LSD),由基因变异导致水解酶α-半乳糖苷酶A(α-GalA)缺乏引起。糖鞘脂球三糖神经酰胺(Gb3)在皮肤、肾脏、大脑、关节、血管壁和眼睛等器官中进行性蓄积,这导致了广泛的临床表现,且往往缺乏特异性。结果,可能会出现具有临床相关性且危及生命的并发症,如恶性室性心律失常、心源性猝死、终末期肾衰竭和中风。在本综述中,我们将描述FD患者的临床特征以及多学科管理的当前观点。