Department of Pharmacology and Physiology, Oklahoma State University College of Osteopathic Medicine, Tahlequah, OK, USA.
Waggoner Center for Alcohol and Addiction Research, The University of Texas at Austin, Austin, TX, USA.
Biochem Pharmacol. 2022 May;199:114993. doi: 10.1016/j.bcp.2022.114993. Epub 2022 Mar 15.
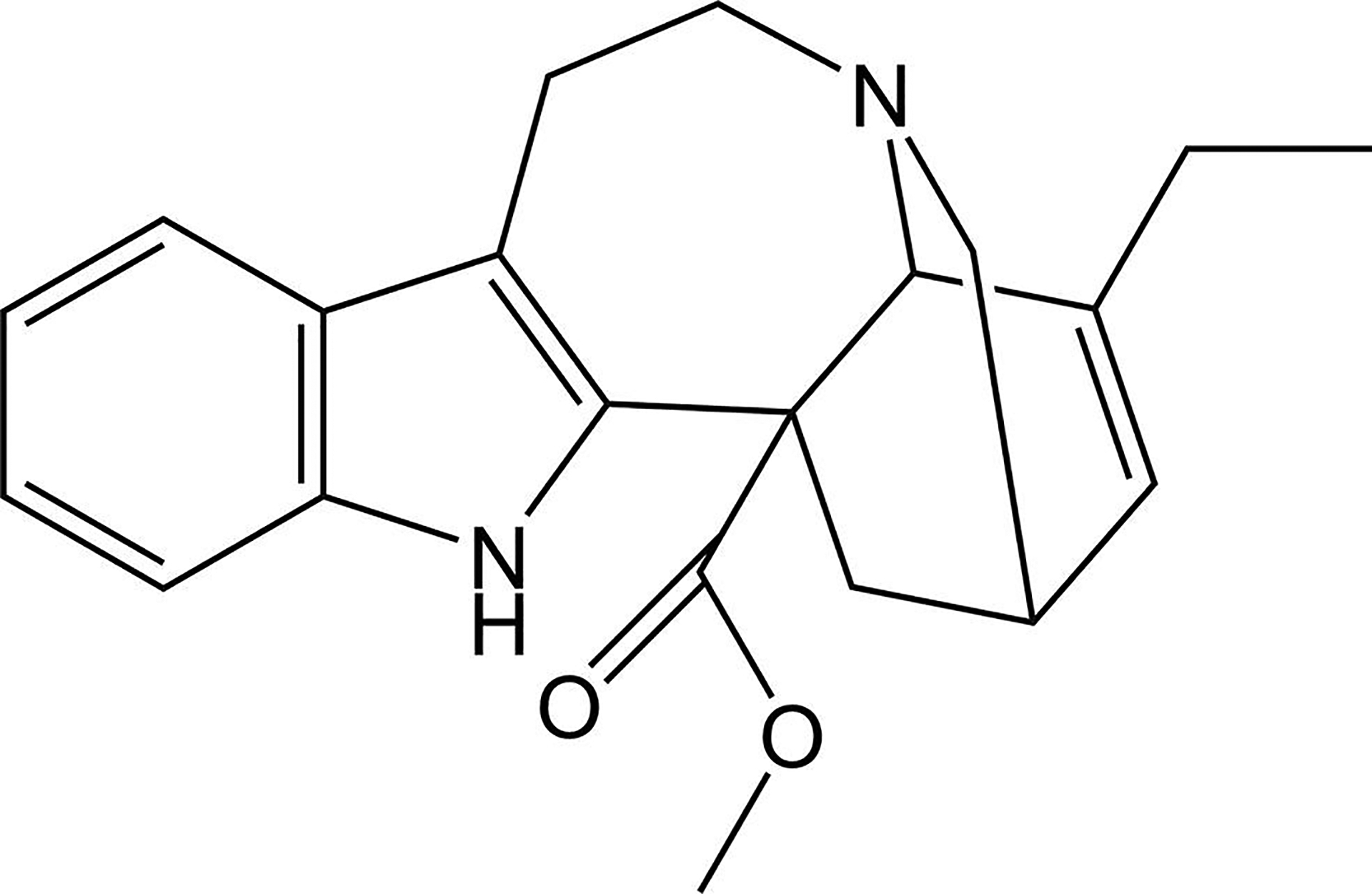
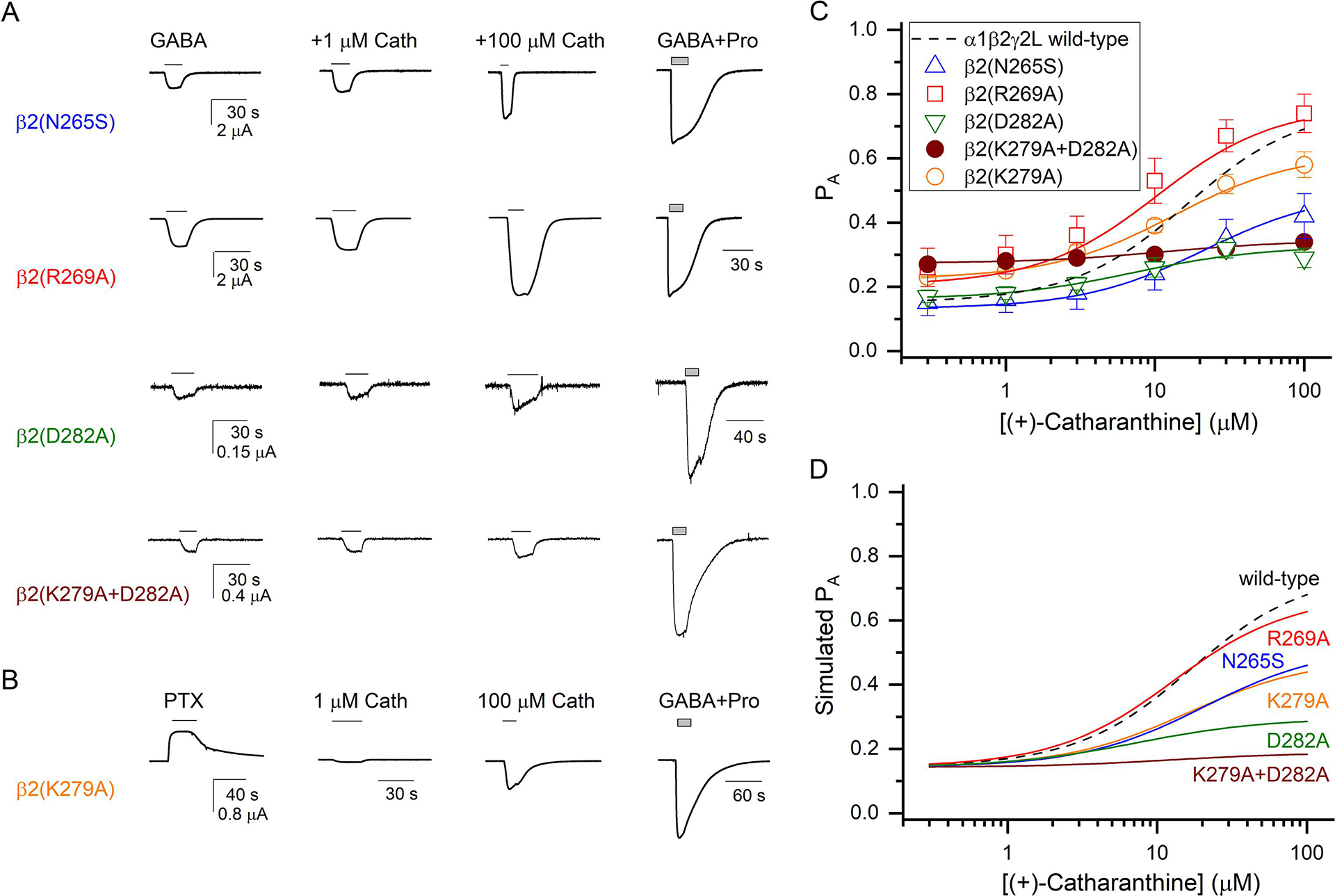
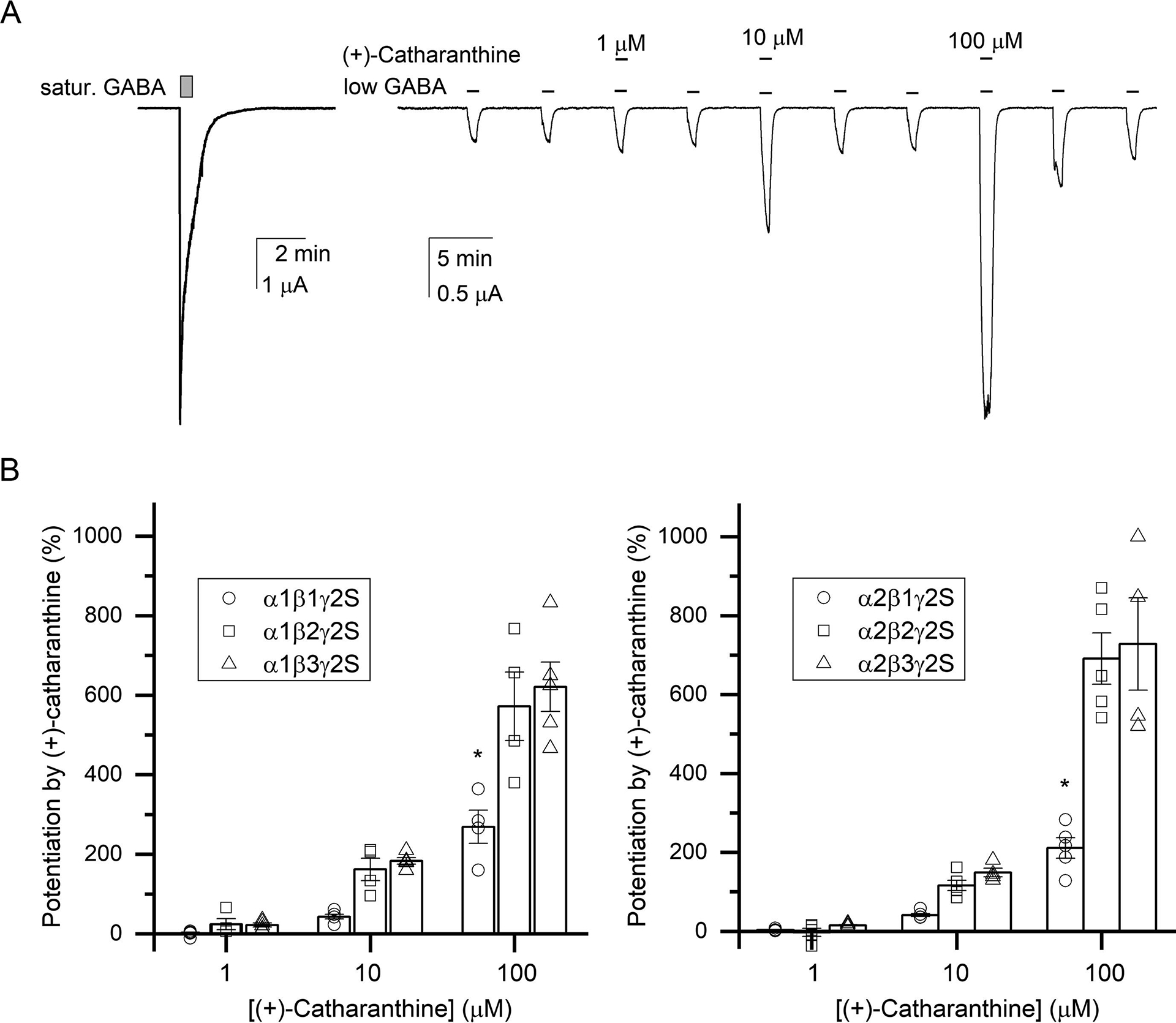
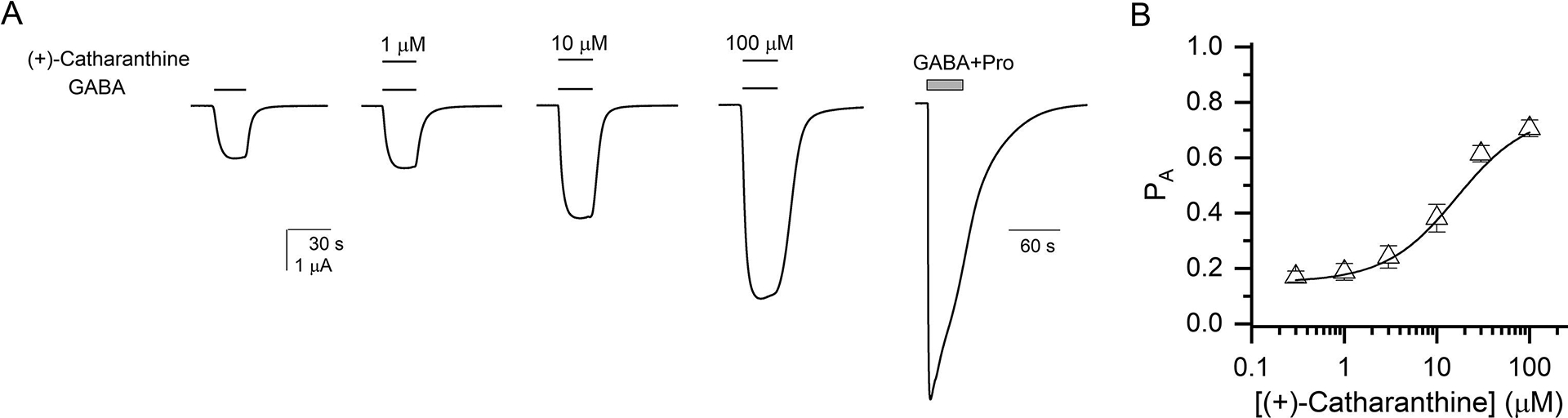
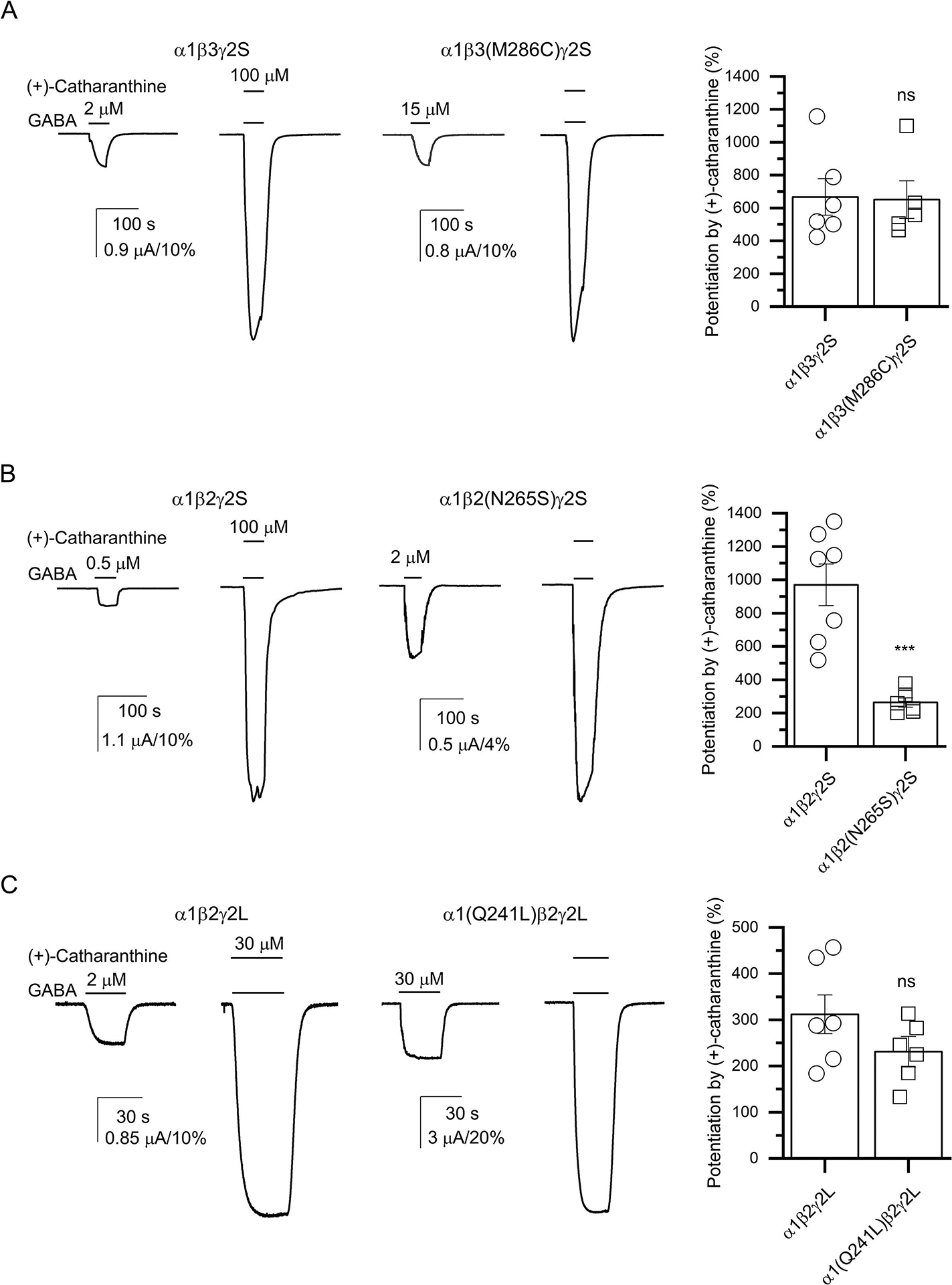
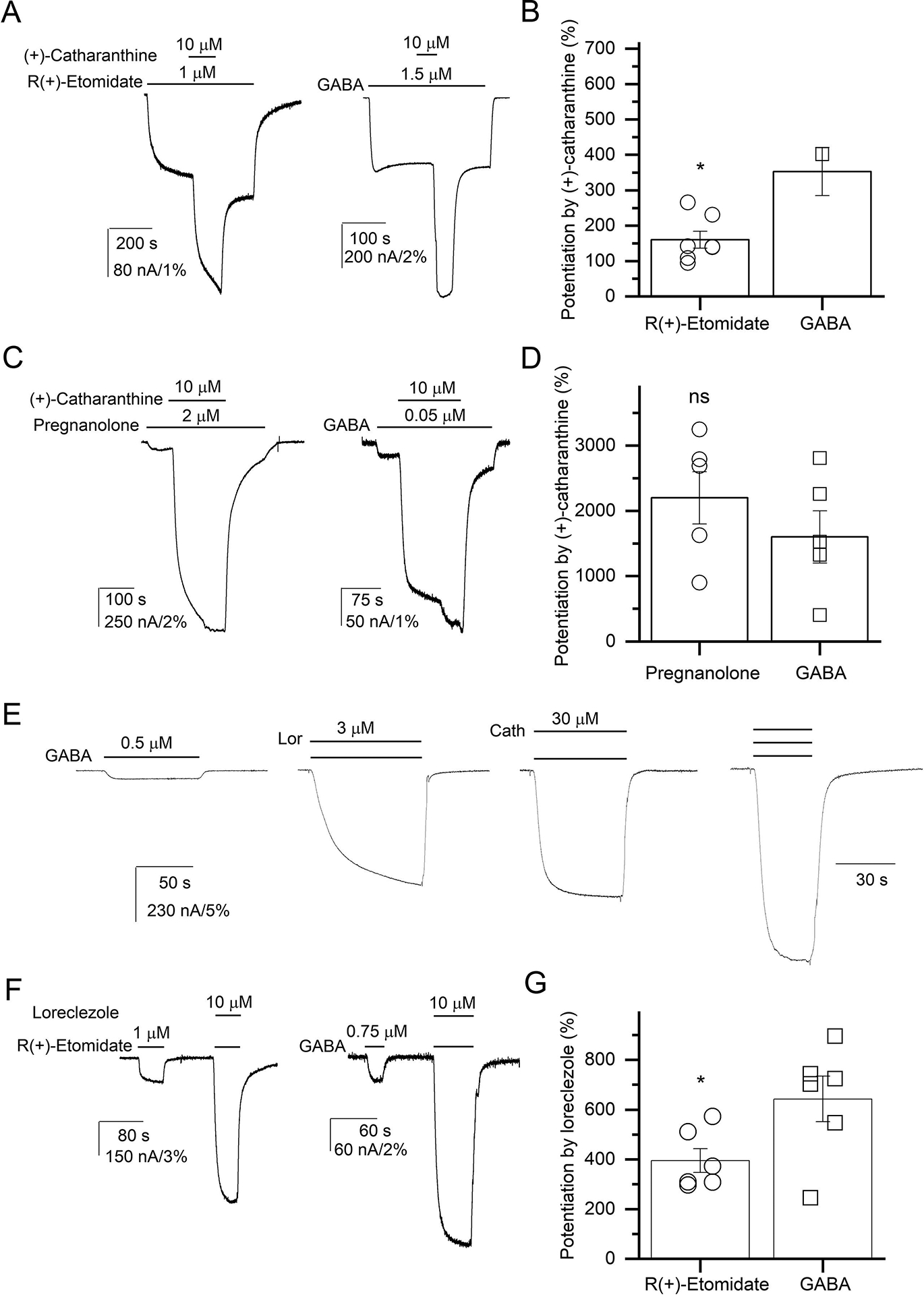
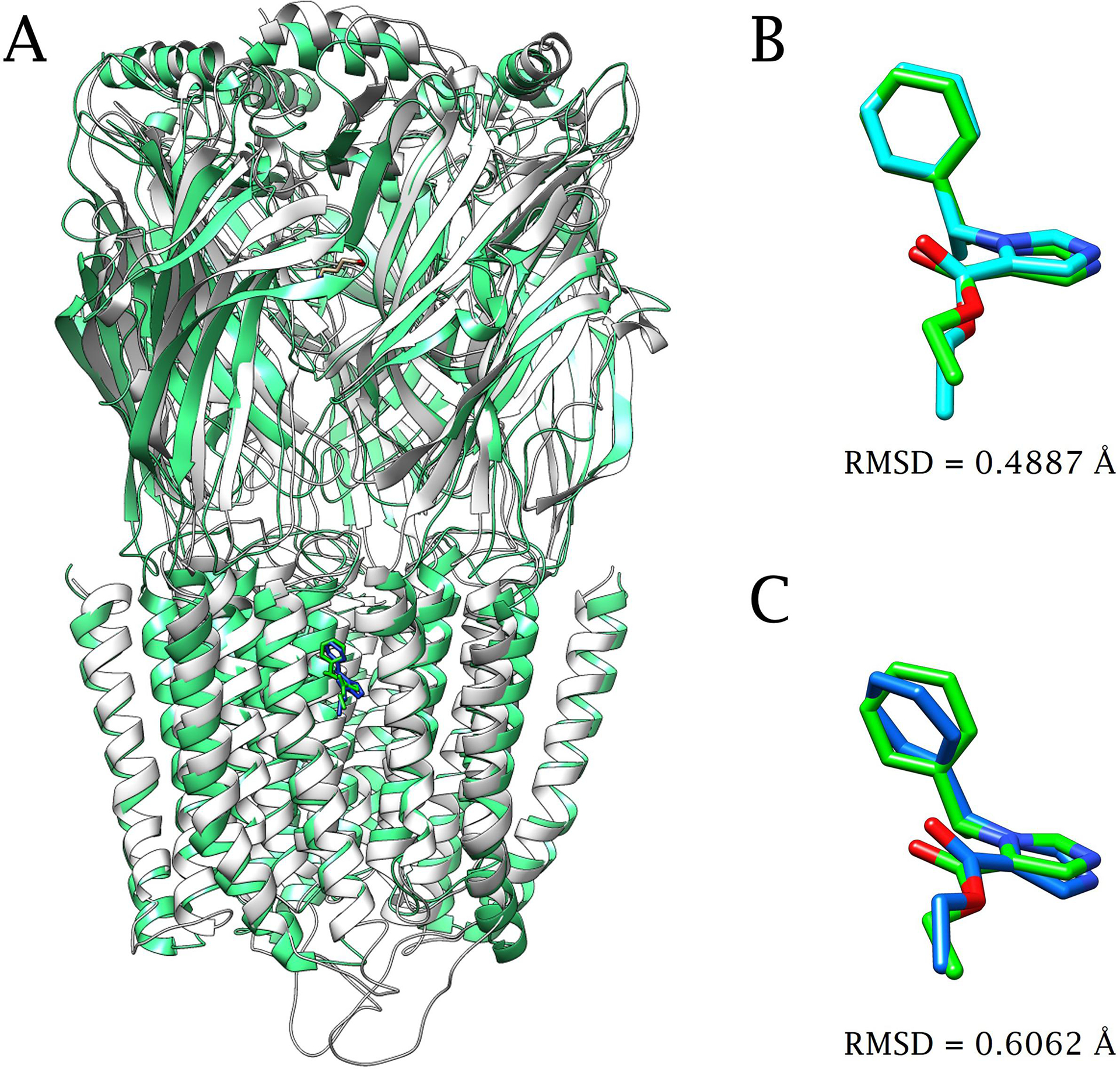
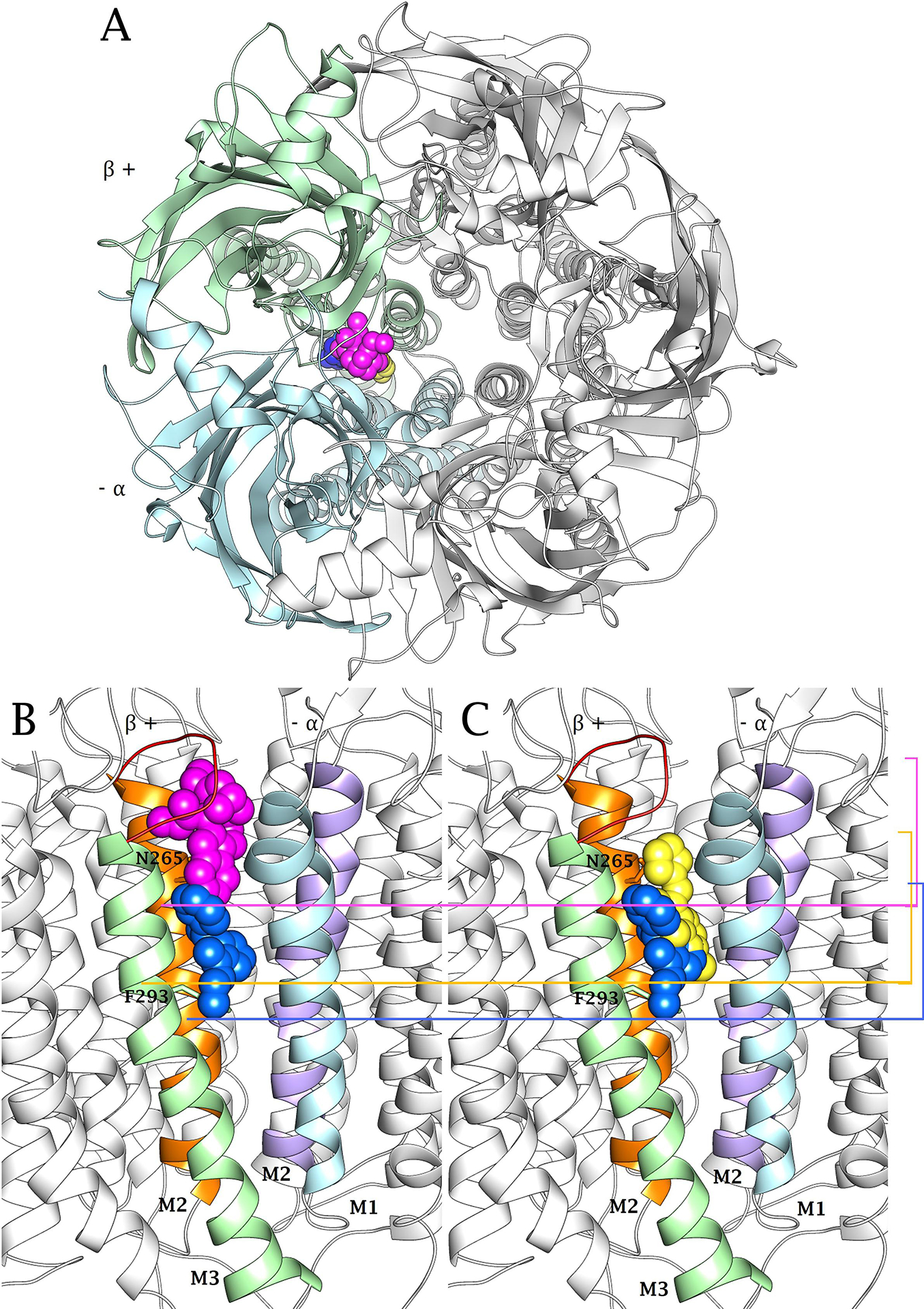
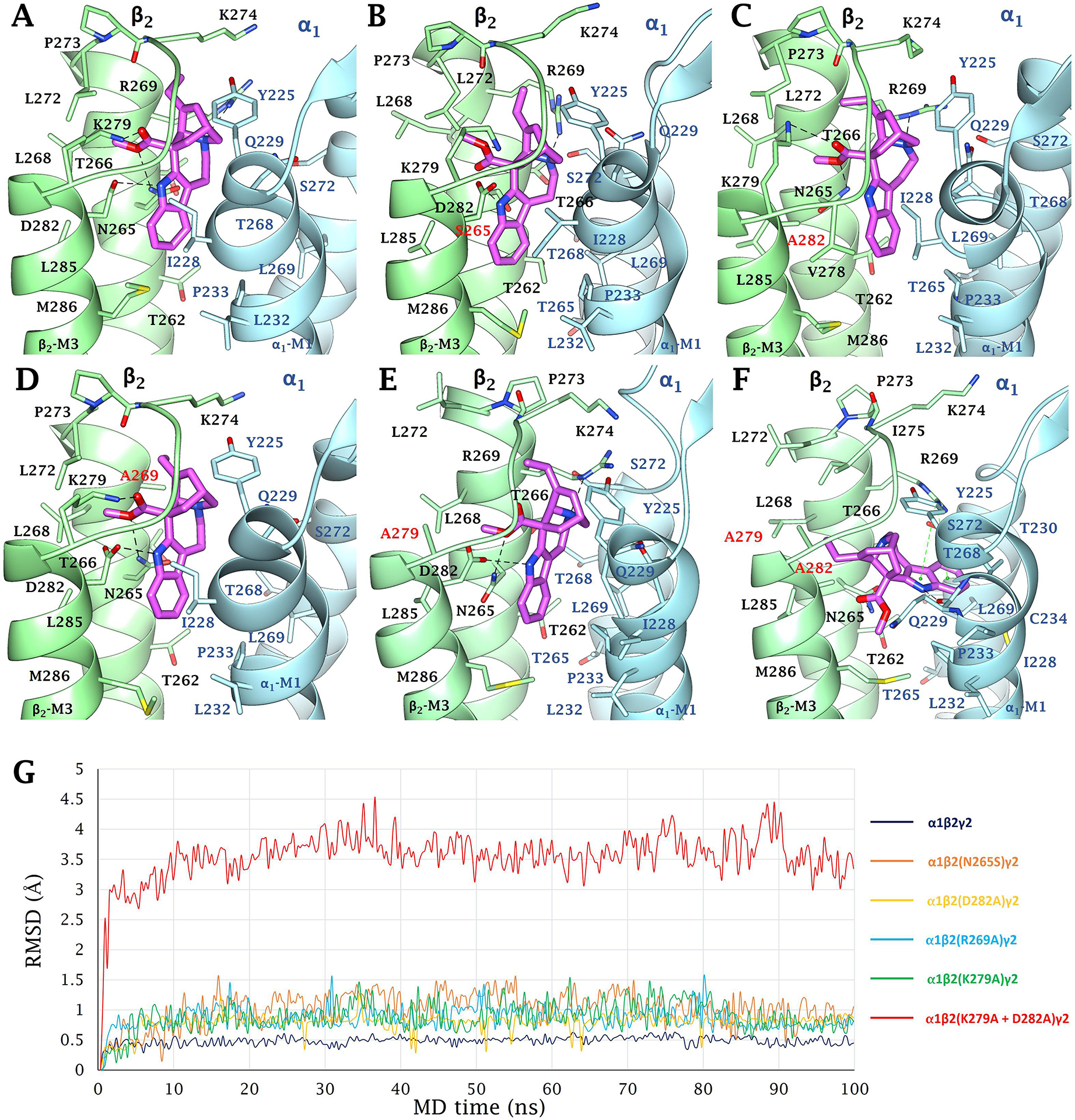
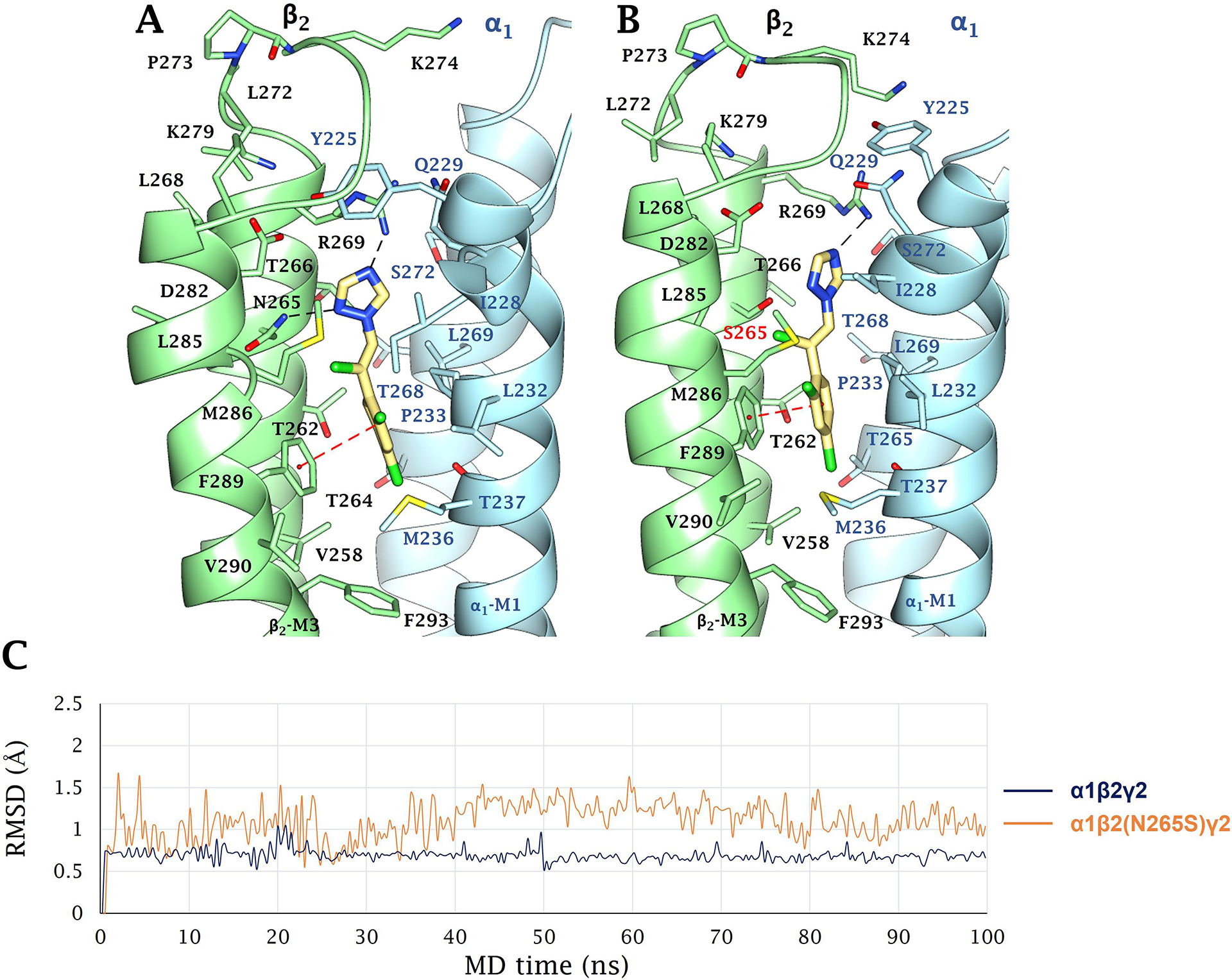
(+)-Catharanthine, a coronaridine congener, potentiates the γ-aminobutyric acid type A receptor (GABAR) and induces sedation through a non-benzodiazepine mechanism, but the specific site of action and intrinsic mechanism have not beendefined. Here, we describe GABAR subtype selectivity and location of the putative binding site for (+)-catharanthine using electrophysiological, site-directed mutagenesis, functional competition, and molecular docking experiments. Electrophysiological and in silico experiments showed that (+)-catharanthine potentiates the responses to low, subsaturating GABA at β2/3-containing GABARs 2.4-3.5 times more efficaciously than at β1-containing GABARs. The activity of (+)-catharanthine is reduced by the β2(N265S) mutation that decreases GABAR potentiation by loreclezole, but not by the β3(M286C) or α1(Q241L) mutations that reduce receptor potentiation by R(+)-etomidate or neurosteroids, respectively. Competitive functional experiments indicated that the binding site for (+)-catharanthine overlaps that for loreclezole, but not those for R(+)-etomidate or potentiating neurosteroids. Molecular docking experiments suggested that (+)-catharanthine binds at the β(+)/α(-) intersubunit interface near the TM2-TM3 loop, where it forms H-bonds with β2-D282 (TM3), β2-K279 (TM2-TM3 loop), and β2-N265 and β2-R269 (TM2). Site-directed mutagenesis experiments supported the in silico results, demonstrating that the K279A and D282A substitutions, that lead to a loss of H-bonding ability of the mutated residue, and the N265S mutation, impair the gating efficacy of (+)-catharanthine. We infer that (+)-catharanthine potentiates the GABAR through several H-bond interactions with a binding site located in the β(+)/α(-) interface in the transmembrane domain, near the TM2-TM3 loop, where it overlaps with loreclezole binding site.
(+)-石蒜裂碱是一种藜芦定的同系物,通过非苯二氮䓬机制增强γ-氨基丁酸 A 型受体(GABAR)并诱导镇静作用,但作用部位和内在机制尚未确定。在这里,我们使用电生理学、定点突变、功能竞争和分子对接实验描述了(+)-石蒜裂碱对 GABAR 亚型的选择性和假定结合位点。电生理学和计算机实验表明,(+)-石蒜裂碱使含有β2/3 的 GABARs 对低浓度、亚饱和 GABA 的反应增强了 2.4-3.5 倍,而对含有β1 的 GABARs 则增强了 2.4-3.5 倍。(+)-石蒜裂碱的活性被β2(N265S)突变降低,该突变降低了 loreclezole 对 GABAR 的增强作用,但不被β3(M286C)或α1(Q241L)突变降低,这分别降低了 R(+)-etomidate 或神经甾体对受体的增强作用。竞争性功能实验表明,(+)-石蒜裂碱的结合位点与 loreclezole 重叠,但不与 R(+)-etomidate 或增强型神经甾体重叠。分子对接实验表明,(+)-石蒜裂碱结合在β(+)/α(-)亚基界面附近的 TM2-TM3 环上,在那里它与β2-D282(TM3)、β2-K279(TM2-TM3 环)和β2-N265 和β2-R269(TM2)形成氢键。定点突变实验支持了计算机模拟结果,表明 K279A 和 D282A 取代导致突变残基失去氢键形成能力,以及 N265S 突变,损害了(+)-石蒜裂碱的门控功效。我们推断,(+)-石蒜裂碱通过与位于跨膜域β(+)/α(-)界面、TM2-TM3 环附近的结合位点的几个氢键相互作用增强 GABAR,该结合位点与 loreclezole 的结合位点重叠。