Ghaleb Youmna, Elbitar Sandy, Philippi Anne, El Khoury Petra, Azar Yara, Andrianirina Miangaly, Loste Alexia, Abou-Khalil Yara, Nicolas Gaël, Le Borgne Marie, Moulin Philippe, Di-Filippo Mathilde, Charrière Sybil, Farnier Michel, Yelnick Cécile, Carreau Valérie, Ferrières Jean, Lecerf Jean-Michel, Derksen Alexa, Bernard Geneviève, Gauthier Marie-Soleil, Coulombe Benoit, Lütjohann Dieter, Fin Bertrand, Boland Anne, Olaso Robert, Deleuze Jean-François, Rabès Jean-Pierre, Boileau Catherine, Abifadel Marianne, Varret Mathilde
INSERM, Laboratory for Vascular Translational Science (LVTS), F-75018 Paris, France.
Laboratory of Biochemistry and Molecular Therapeutics (LBTM), Faculty of Pharmacy, Pôle Technologie-Santé (PTS), Saint-Joseph University, Beirut 1004 2020, Lebanon.
Metabolites. 2022 Mar 18;12(3):262. doi: 10.3390/metabo12030262.
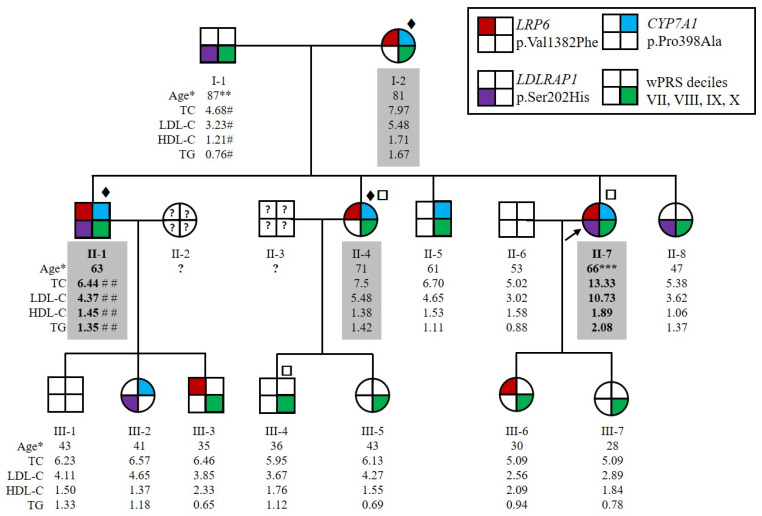
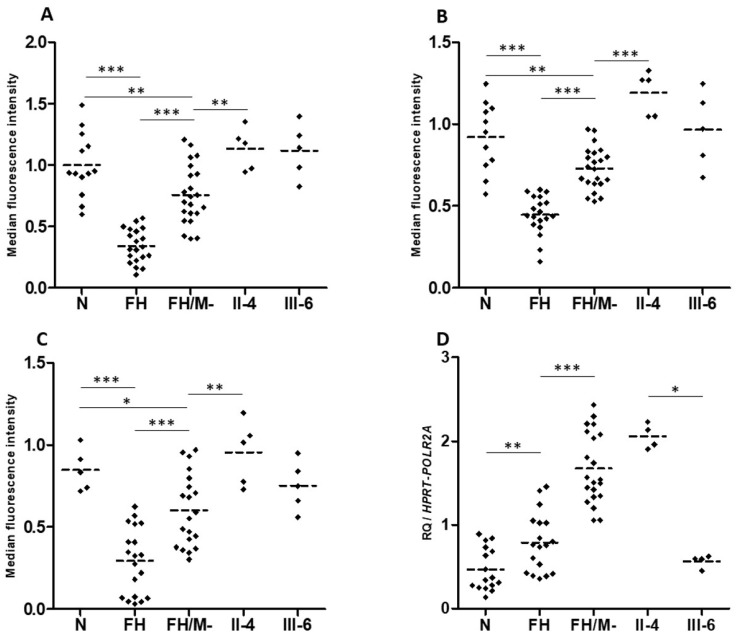
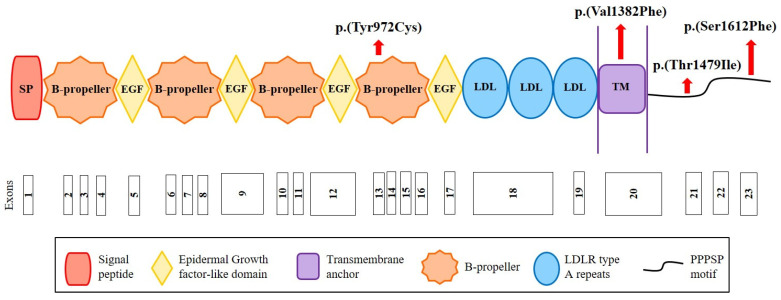
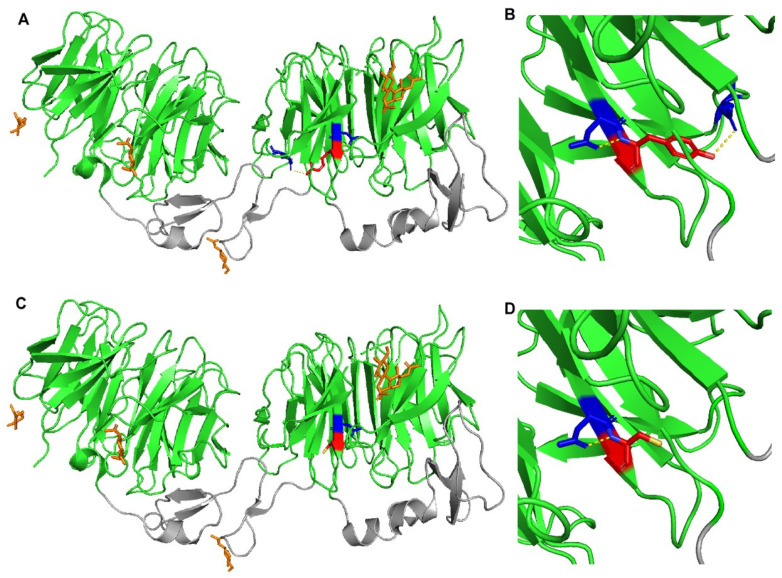
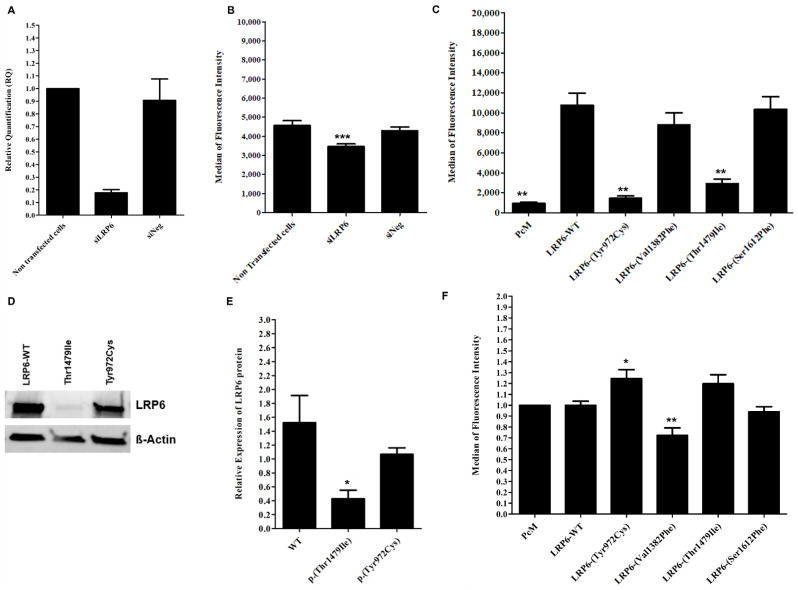
Autosomal Dominant Hypercholesterolemia (ADH) is a genetic disorder caused by pathogenic variants in , , and genes. We sought to identify new candidate genes responsible for the ADH phenotype in patients without pathogenic variants in the known ADH-causing genes by focusing on a French family with affected and non-affected members who presented a high ADH polygenic risk score (wPRS). Linkage analysis, whole exome and whole genome sequencing resulted in the identification of variants p.(Pro398Ala) in , p.(Val1382Phe) in and p.(Ser202His) in . A total of 6 other variants were identified in 6 of 160 unrelated ADH probands: p.(Ala13Val) and p.(Aps347Asn) in ; p.(Tyr972Cys), p.(Thr1479Ile) and p.(Ser1612Phe) in ; and p.(Ser202LeufsTer19) in . All six probands presented a moderate wPRS. Serum analyses of carriers of the p.(Pro398Ala) variant in showed no differences in the synthesis of bile acids compared to the serums of non-carriers. Functional studies of the four mutants in HEK293T cells resulted in contradictory results excluding a major effect of each variant alone. Within the family, none of the heterozygous for only the p.(Ser202His) variant presented ADH. Altogether, each variant individually does not result in elevated LDL-C; however, the oligogenic combination of two or three variants reveals the ADH phenotype.
常染色体显性高胆固醇血症(ADH)是一种由 、 、 和 基因中的致病变异引起的遗传疾病。我们试图通过关注一个有患病和未患病成员且具有高ADH多基因风险评分(wPRS)的法国家庭,来确定在已知导致ADH的基因中没有致病变异的患者中导致ADH表型的新候选基因。连锁分析、全外显子组和全基因组测序导致在 中鉴定出p.(Pro398Ala)变异、在 中鉴定出p.(Val1382Phe)变异以及在 中鉴定出p.(Ser202His)变异。在160名无关的ADH先证者中的6名中总共鉴定出了6个其他变异:在 中有p.(Ala13Val)和p.(Aps347Asn);在 中有p.(Tyr972Cys)、p.(Thr1479Ile)和p.(Ser1612Phe);在 中有p.(Ser202LeufsTer19)。所有6名先证者都表现出中等的wPRS。对 中p.(Pro398Ala)变异携带者的血清分析表明,与非携带者的血清相比,胆汁酸合成没有差异。对HEK293T细胞中四个 突变体的功能研究得出了相互矛盾的结果,排除了每个变异单独产生的主要影响。在这个家族中,仅 中p.(Ser202His)变异的杂合子中没有一个表现出ADH。总之,每个变异单独都不会导致低密度脂蛋白胆固醇(LDL-C)升高;然而,两个或三个变异的寡基因组合揭示了ADH表型。