Division of Neurology, Department of Pediatrics, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX, 75390, USA.
Present affiliation: Corteva Agriscience, Johnston, IA, 50131, USA.
Neurotherapeutics. 2022 Apr;19(3):982-993. doi: 10.1007/s13311-022-01218-7. Epub 2022 Mar 28.
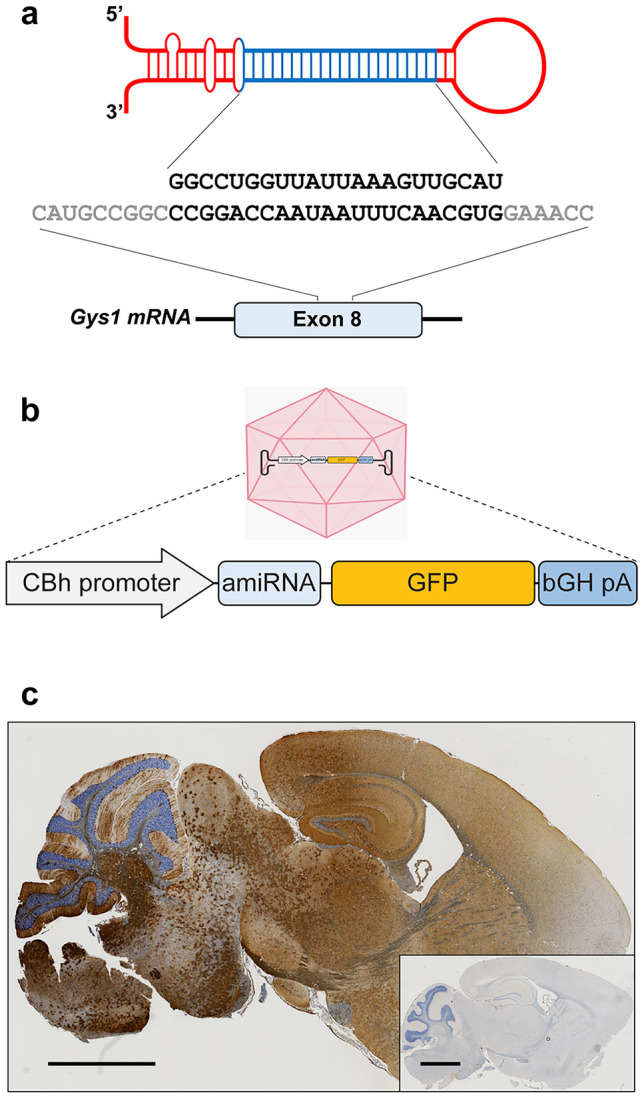
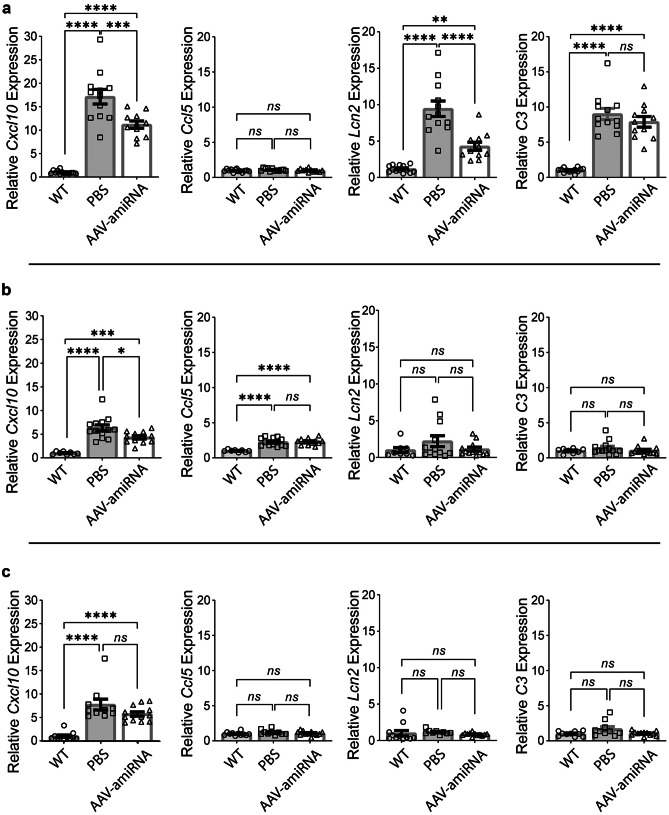
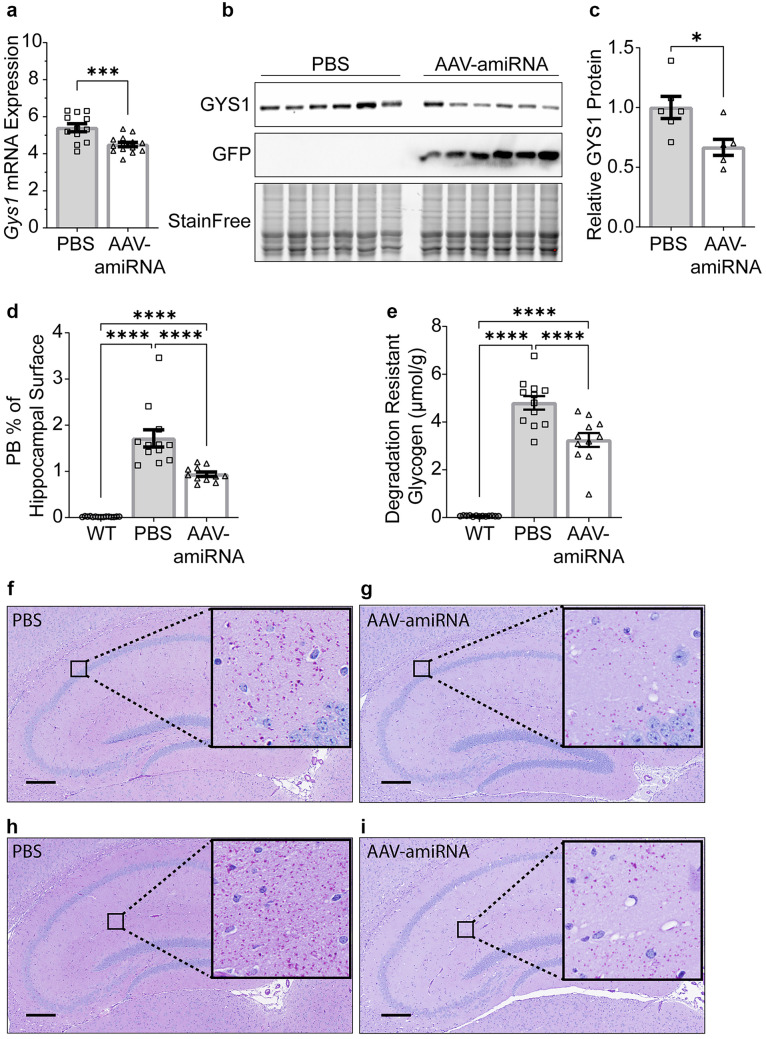
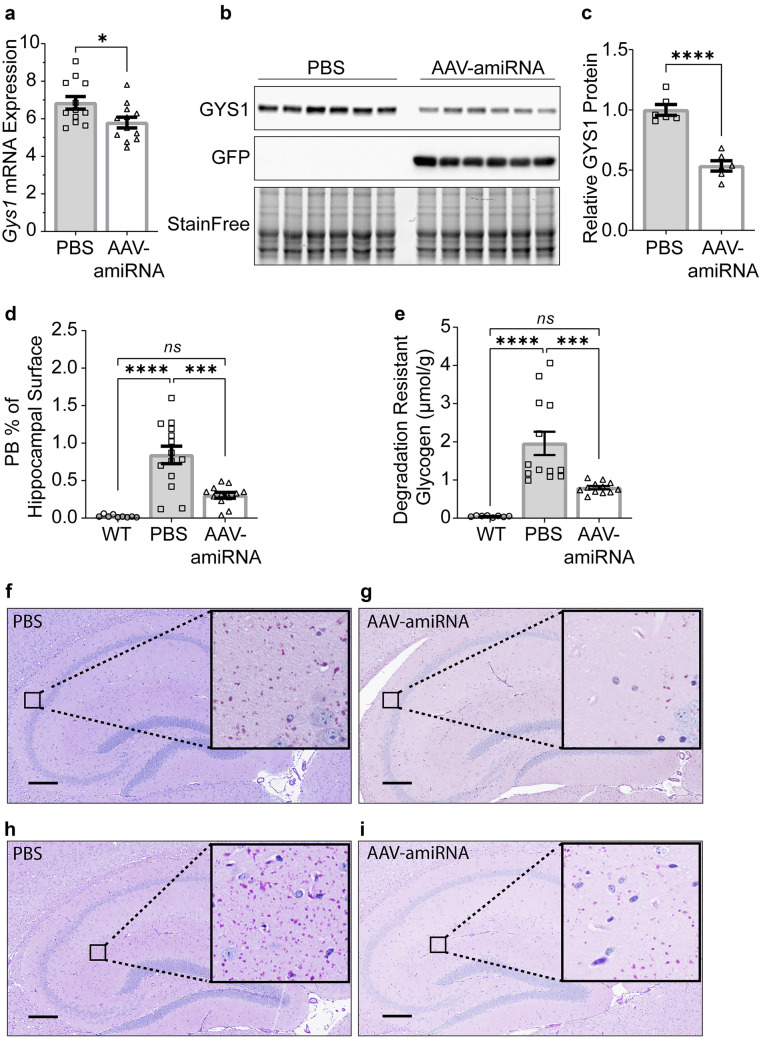
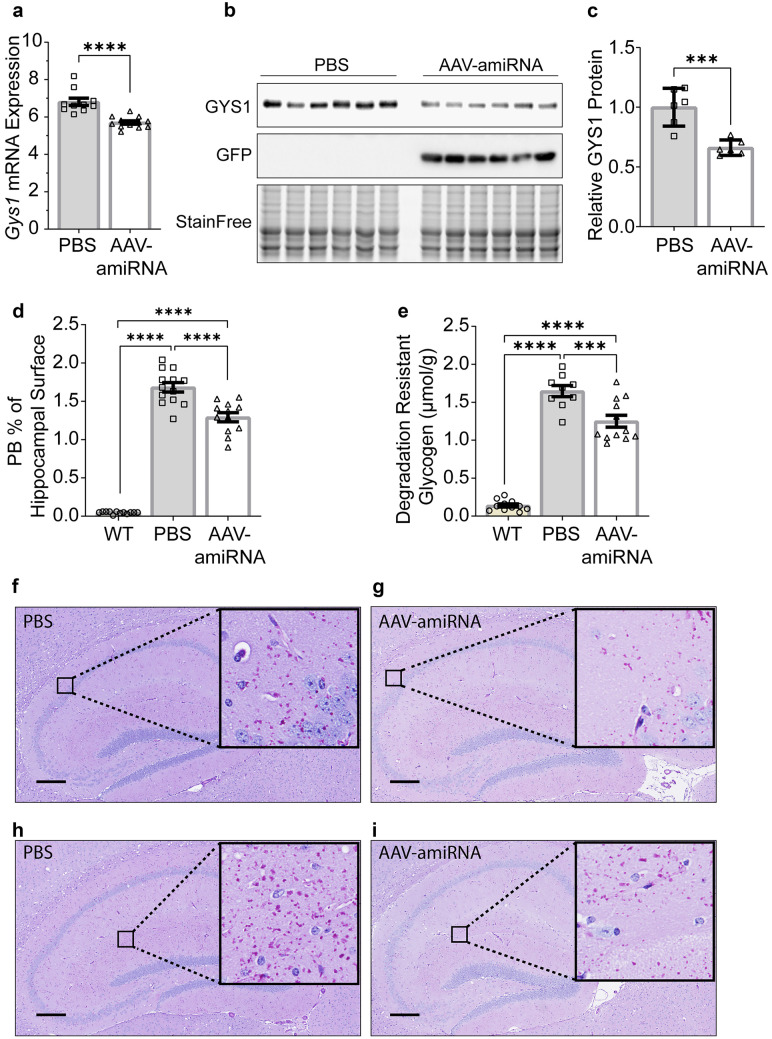
Adult polyglucosan body disease (APBD) and Lafora disease (LD) are autosomal recessive glycogen storage neurological disorders. APBD is caused by mutations in the glycogen branching enzyme (GBE1) gene and is characterized by progressive upper and lower motor neuron dysfunction and premature death. LD is a fatal progressive myoclonus epilepsy caused by loss of function mutations in the EPM2A or EPM2B gene. These clinically distinct neurogenetic diseases share a common pathology. This consists of time-dependent formation, precipitation, and accumulation of an abnormal form of glycogen (polyglucosan) into gradually enlarging inclusions, polyglucosan bodies (PBs) in ever-increasing numbers of neurons and astrocytes. The growth and spread of PBs are followed by astrogliosis, microgliosis, and neurodegeneration. The key defect in polyglucosans is that their glucan branches are longer than those of normal glycogen, which prevents them from remaining in solution. Since the lengths of glycogen branches are determined by the enzyme glycogen synthase, we hypothesized that downregulating this enzyme could prevent or hinder the generation of the pathogenic PBs. Here, we pursued an adeno-associated virus vector (AAV) mediated RNA-interference (RNAi) strategy. This approach resulted in approximately 15% reduction of glycogen synthase mRNA and an approximately 40% reduction of PBs across the brain in the APBD and both LD mouse models. This was accompanied by improvements in early neuroinflammatory markers of disease. This work represents proof of principle toward developing a single lifetime dose therapy for two fatal neurological diseases: APBD and LD. The approach is likely applicable to other severe and common diseases of glycogen storage.
成人多聚葡萄糖体病 (APBD) 和拉福拉病 (LD) 是常染色体隐性遗传的糖原贮积神经系统疾病。APBD 是由糖原分支酶 (GBE1) 基因突变引起的,其特征是进行性上下运动神经元功能障碍和过早死亡。LD 是一种致命的进行性肌阵挛性癫痫,由 EPM2A 或 EPM2B 基因突变导致功能丧失引起。这些临床表现不同的神经遗传疾病具有共同的病理学特征。这包括异常形式的糖原(多聚葡萄糖)随时间形成、沉淀和积累,逐渐增大的包涵体、神经元和星形胶质细胞中越来越多的多聚葡萄糖体 (PBs)。PB 的生长和扩散随后伴随着星形胶质细胞增生、小胶质细胞增生和神经退行性变。多聚葡萄糖的关键缺陷是其葡聚糖分支比正常糖原长,这阻止了它们保持在溶液中。由于糖原分支的长度由酶糖原合酶决定,我们假设下调这种酶可以防止或阻碍致病性 PBs 的产生。在这里,我们采用了腺相关病毒载体 (AAV) 介导的 RNA 干扰 (RNAi) 策略。这种方法导致 APBD 和两种 LD 小鼠模型的大脑中糖原合酶 mRNA 减少约 15%,PB 减少约 40%。这伴随着疾病早期神经炎症标志物的改善。这项工作证明了开发一种针对两种致命神经疾病(APBD 和 LD)的单一终身剂量治疗方法的原理。这种方法可能适用于其他严重和常见的糖原贮积疾病。