Department of Physiology, College of Basic Medical Sciences, Liaoning Provincial Key Laboratory of Cerebral Diseases, National-Local Joint Engineering Research Center for Drug-Research and Development (R&D) of Neurodegenerative Diseases, Dalian Medical University, Dalian, China.
Department of Neurology and Clinical Research Center of Neurological Disease, The Second Affiliated Hospital of Soochow University, Suzhou, China.
Aging Cell. 2022 May;21(5):e13593. doi: 10.1111/acel.13593. Epub 2022 Mar 30.
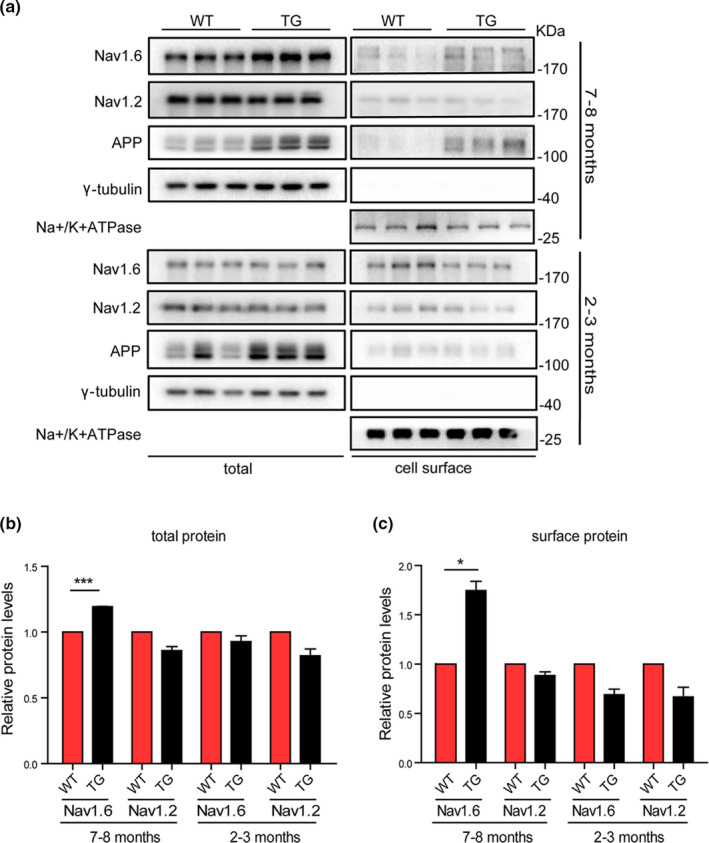
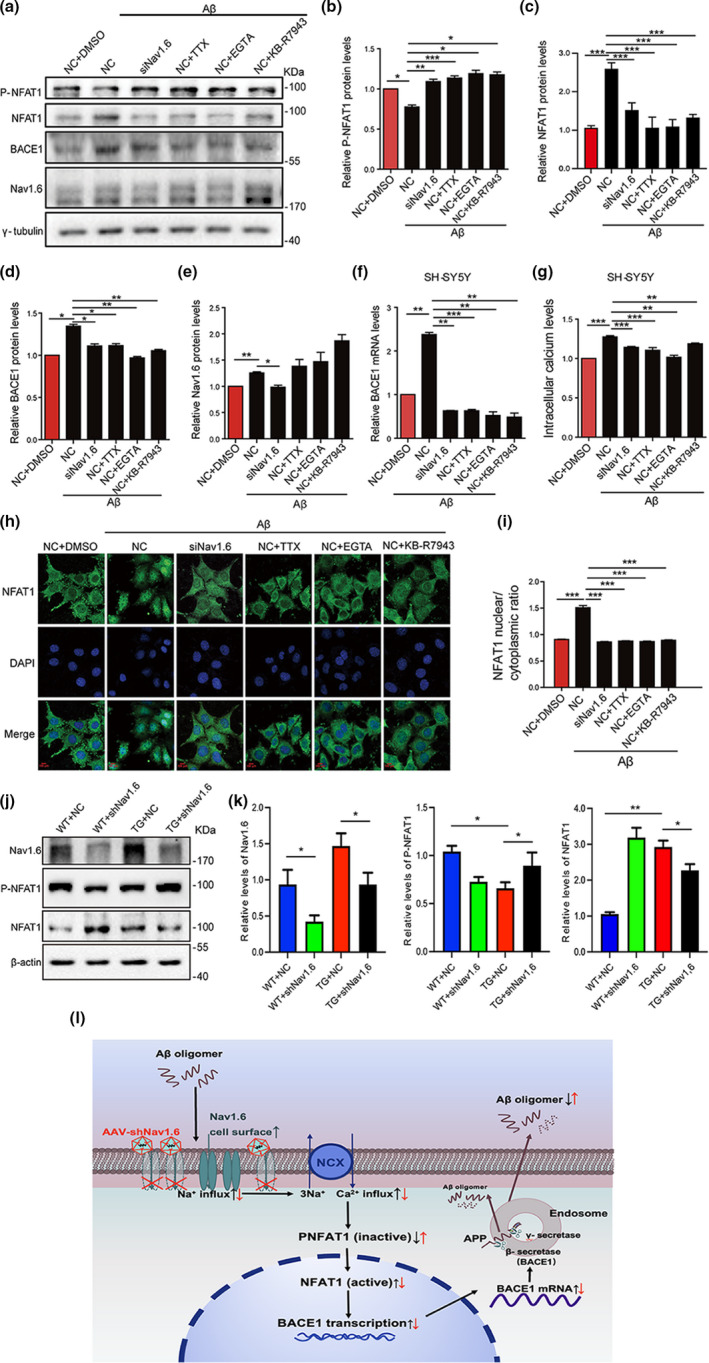
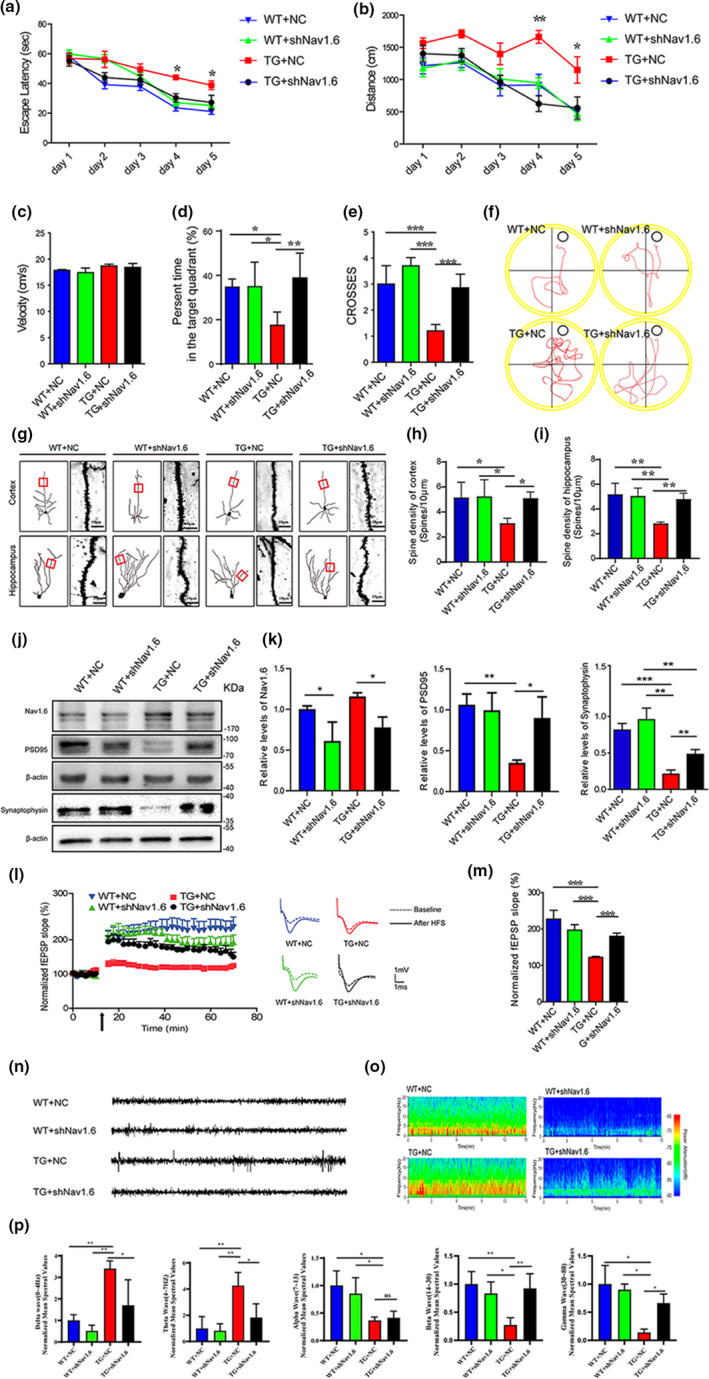
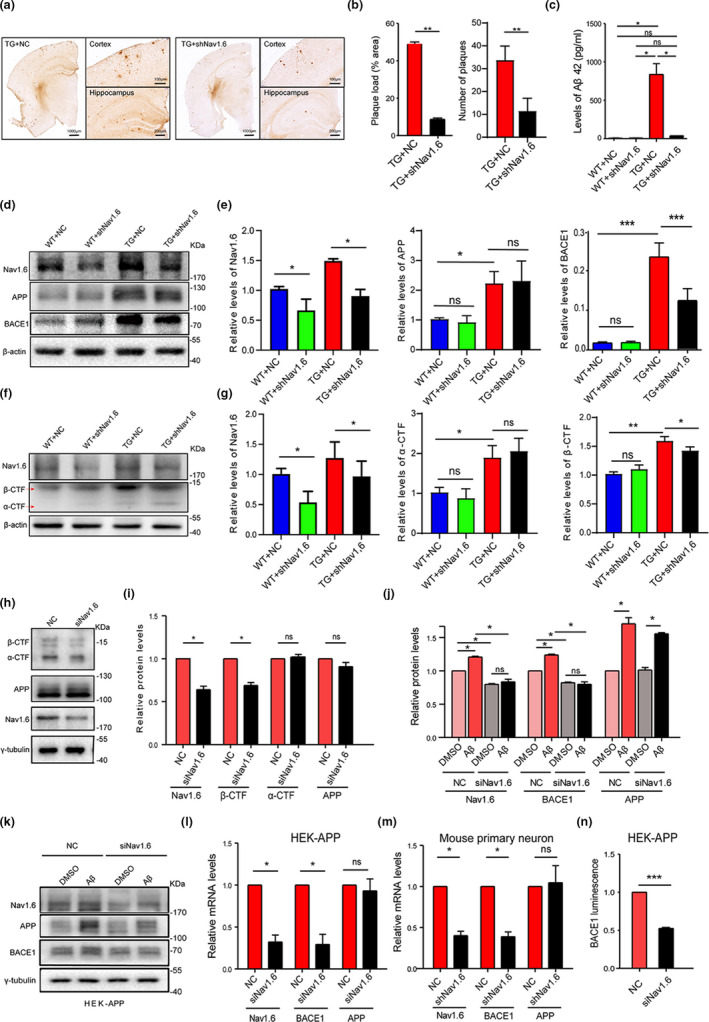
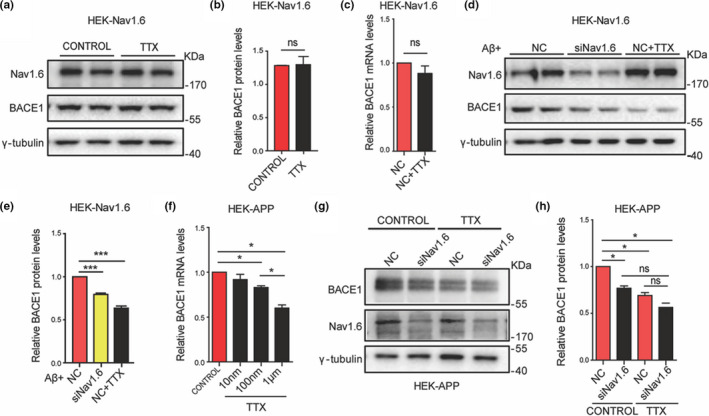
Aberrant increases in neuronal network excitability may contribute to cognitive deficits in Alzheimer's disease (AD). However, the mechanisms underlying hyperexcitability of neurons are not fully understood. Voltage-gated sodium channels (VGSC or Nav), which are involved in the formation of excitable cell's action potential and can directly influence the excitability of neural networks, have been implicated in AD-related abnormal neuronal hyperactivity and higher incidence of spontaneous non-convulsive seizures. Here, we have shown that the reduction of VGSC α-subunit Nav1.6 (by injecting adeno-associated virus (AAV) with short hairpin RNA (shRNA) into the hippocampus) rescues cognitive impairments and attenuates synaptic deficits in APP/PS1 transgenic mice. Concurrently, amyloid plaques in the hippocampus and levels of soluble Aβ are significantly reduced. Interfering with Nav1.6 reduces the transcription level of β-site APP-cleaving enzyme 1 (BACE1), which is Aβ-dependent. In the presence of Aβ oligomers, knockdown of Nav1.6 reduces intracellular calcium overload by suppressing reverse sodium-calcium exchange channel, consequently increasing inactive NFAT1 (the nuclear factor of activated T cells) levels and thus reducing BACE1 transcription. This mechanism leads to a reduction in the levels of Aβ in APP/PS1 transgenic mice, alleviates synaptic loss, improves learning and memory disorders in APP/PS1 mice after downregulating Nav1.6 in the hippocampus. Our study offers a new potential therapeutic strategy to counteract hippocampal hyperexcitability and subsequently rescue cognitive deficits in AD by selective blockade of Nav1.6 overexpression and/or hyperactivity.
神经元网络兴奋性的异常增加可能导致阿尔茨海默病(AD)的认知障碍。然而,神经元过度兴奋的机制尚未完全阐明。电压门控钠离子通道(VGSC 或 Nav)参与可兴奋细胞动作电位的形成,并能直接影响神经网络的兴奋性,与 AD 相关的神经元异常兴奋和自发性非惊厥性癫痫发作的发生率较高有关。在这里,我们已经表明,通过向海马体注射含有短发夹 RNA(shRNA)的腺相关病毒(AAV),减少 VGSCα 亚基 Nav1.6(Nav1.6)的表达,可以挽救 APP/PS1 转基因小鼠的认知障碍并减轻突触缺陷。同时,海马体中的淀粉样斑块和可溶性 Aβ 的水平显著降低。干扰 Nav1.6 会降低β-位淀粉样前体蛋白裂解酶 1(BACE1)的转录水平,BACE1 是 Aβ 依赖性的。在 Aβ 寡聚物存在的情况下,Nav1.6 的敲低通过抑制反向钠钙交换通道来减少细胞内钙超载,从而增加失活的 NFAT1(激活 T 细胞的核因子)水平,从而减少 BACE1 的转录。这种机制导致 APP/PS1 转基因小鼠中 Aβ 水平降低,减轻突触丢失,改善 APP/PS1 小鼠的学习和记忆障碍,其机制是下调 Nav1.6 后,海马体中 Nav1.6 的表达下调。我们的研究为通过选择性阻断 Nav1.6 的过度表达和/或过度活跃来对抗海马体过度兴奋并随后挽救 AD 中的认知障碍提供了一种新的潜在治疗策略。