Department of Hematogenetics, National Institute of Immunohematology, Indian Council of Medical Research [ICMR], 13th Floor, K.E.M. Hospital Campus, Parel, Mumbai, 400 012, India.
Sci Rep. 2022 Mar 30;12(1):5414. doi: 10.1038/s41598-022-09250-5.
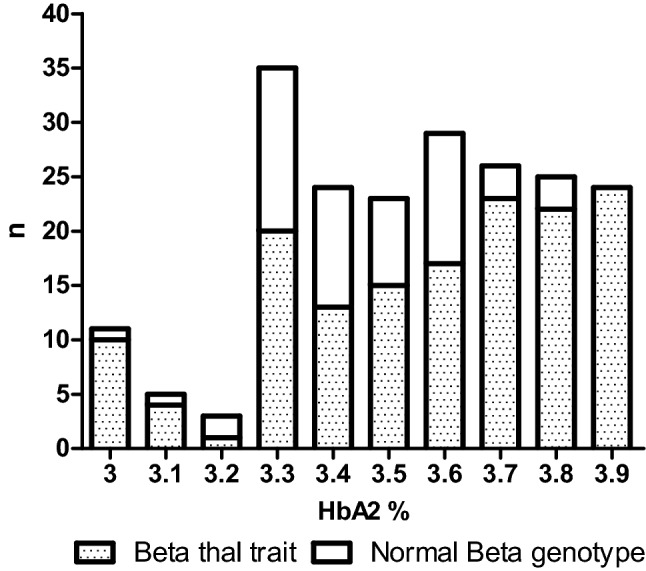
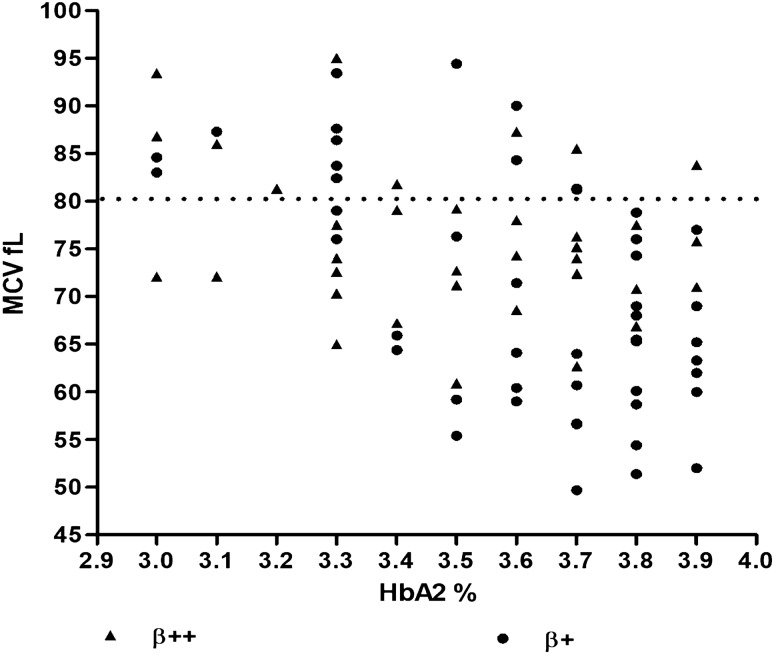
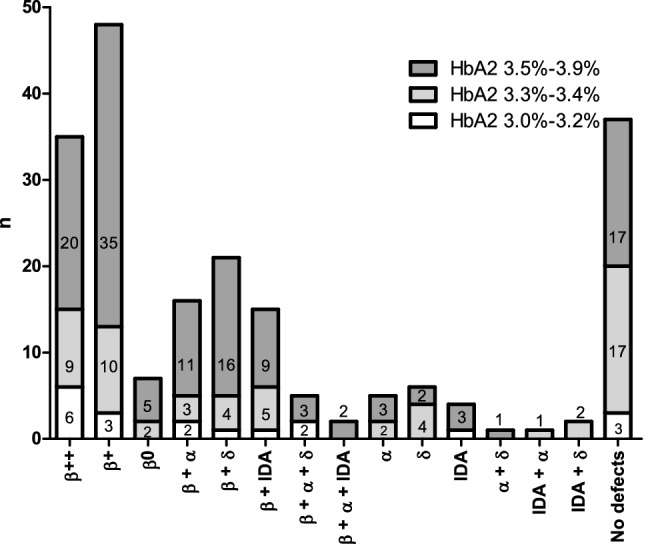
Increased HbA levels are the characteristic feature of β-thalassemia carriers. A subset of carriers however do not show HbA levels in the typical carrier range (≥ 4.0%) but show borderline HbA levels. As a result, these carriers escape diagnosis and carry the risk of having β-thalassemia major offspring. Borderline HbA values may occur as a consequence of mild β-thalassemia mutations, co-inherited β-thalassemia and α- or δ- thalassemia or iron deficiency anemia. However, there is insufficient knowledge regarding the cause of borderline HbA levels in specific populations. This study aimed to identify the determinants of borderline HbA levels (which we have considered as HbA 3.0-3.9%) in 205 individuals. Primary screening involved detecting the presence of iron deficiency anemia followed by molecular analysis of α, β and δ globin genes. Remarkably, 168 of 205 individuals were positive for a defect. 87% (149/168) of positive individuals were heterozygous for β thalassemia with (59/149) or without (90/149) the presence of co-existing IDA, α or δ gene defects. Notably, 20 of 149 β thalassemia carriers showed HbA < 3.5% and MCV > 80fL. 7 of these 20 carriers were married to carriers of hemoglobinopathies. Our findings describe the genetic basis of borderline HbA levels and emphasize the necessity of a molecular diagnosis in these individuals in the routine clinical setting.
血红蛋白 A(HbA)水平升高是β-地中海贫血携带者的特征。然而,有一部分携带者的 HbA 水平不在典型携带者范围内(≥4.0%),而是处于边缘 HbA 水平。因此,这些携带者会被漏诊,并有可能生育β-地中海贫血重型患儿。边缘 HbA 值可能是由于轻度β-地中海贫血突变、共遗传的β-地中海贫血和α-或δ-地中海贫血或缺铁性贫血引起的。然而,对于特定人群中边缘 HbA 水平的原因,我们的了解还不够充分。本研究旨在确定 205 名个体中边缘 HbA 水平(我们将其视为 3.0-3.9%的 HbA)的决定因素。初步筛查包括检测缺铁性贫血的存在,然后对α、β和δ珠蛋白基因进行分子分析。值得注意的是,205 名个体中有 168 名存在缺陷。168 名阳性个体中有 87%(149/168)为β地中海贫血杂合子,其中(59/149)或不伴有(90/149)同时存在 IDA、α 或δ 基因缺陷。值得注意的是,149 名β地中海贫血携带者中有 20 名 HbA<3.5%且 MCV>80fL。这 20 名携带者中有 7 名与血红蛋白病携带者结婚。我们的研究结果描述了边缘 HbA 水平的遗传基础,并强调了在常规临床环境中对这些个体进行分子诊断的必要性。