Zheng Wei, Fan Dongsheng
Department of Neurology, Peking University Third Hospital, Beijing, China.
Beijing Key Laboratory of Biomarker and Translational Research in Neurodegenerative Diseases, Beijing, China.
Front Aging Neurosci. 2022 Mar 23;14:851135. doi: 10.3389/fnagi.2022.851135. eCollection 2022.
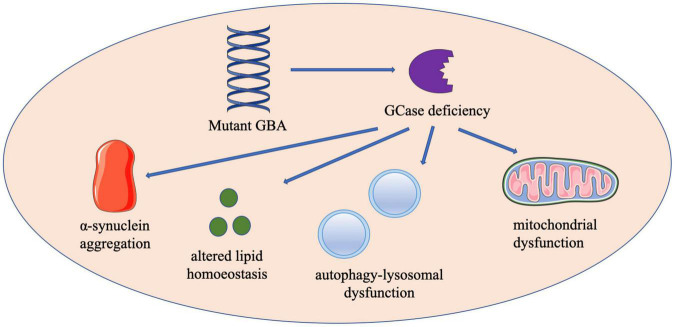
Parkinson's disease (PD) is the second most common neurodegenerative disease and is characterized by multiple motor and non-motor symptoms. Mutations in the glucocerebrosidase () gene, which encodes the lysosomal enzyme glucocerebrosidase (GCase), which hydrolyzes glucosylceramide (GlcCer) to glucose and ceramide, are the most important and common genetic PD risk factors discovered to date. Homozygous mutations result in the most common lysosomal storage disorder, Gaucher's disease (GD), which is classified according to the presence (neuronopathic types, type 2 and 3 GD) or absence (non-neuronopathic type, type 1 GD) of neurological symptoms. The clinical manifestations of PD in patients with mutations are indistinguishable from those of sporadic PD at the individual level. However, accumulating data have indicated that -associated PD patients exhibit a younger age of onset and a greater risk for cognitive impairment and psychiatric symptoms. The mechanisms underlying the increased risk of developing PD in mutant carriers are currently unclear. Contributors to -PD pathogenesis may include mitochondrial dysfunction, autophagy-lysosomal dysfunction, altered lipid homeostasis and enhanced α-synuclein aggregation. Therapeutic strategies for PD and GD targeting mutant GCase mainly include enzyme replacement, substrate reduction, gene and pharmacological small-molecule chaperones. Emerging clinical, genetic and pathogenic studies on mutations and PD are making significant contributions to our understanding of PD-associated pathogenetic pathways, and further elucidating the interactions between GCase activity and neurodegeneration may improve therapeutic approaches for slowing PD progression.
帕金森病(PD)是第二常见的神经退行性疾病,其特征为多种运动和非运动症状。葡萄糖脑苷脂酶()基因发生突变,该基因编码溶酶体酶葡萄糖脑苷脂酶(GCase),GCase可将葡萄糖神经酰胺(GlcCer)水解为葡萄糖和神经酰胺,这是迄今为止发现的最重要且最常见的遗传性PD风险因素。纯合突变会导致最常见的溶酶体贮积症——戈谢病(GD),GD根据是否存在神经症状分为有神经症状(神经病变型,2型和3型GD)或无神经症状(非神经病变型,1型GD)。携带突变的患者中PD的临床表现与散发性PD在个体水平上难以区分。然而,越来越多的数据表明,携带相关突变的PD患者发病年龄较轻,发生认知障碍和精神症状的风险更高。目前尚不清楚携带突变的个体患PD风险增加的潜在机制。与 -PD发病机制相关的因素可能包括线粒体功能障碍、自噬-溶酶体功能障碍、脂质稳态改变以及α-突触核蛋白聚集增强。针对突变型GCase的PD和GD治疗策略主要包括酶替代、底物减少、基因治疗和药理学小分子伴侣。关于突变与PD的新兴临床、遗传和发病机制研究,正在为我们理解与PD相关的发病途径做出重大贡献,进一步阐明GCase活性与神经退行性变之间的相互作用可能会改善减缓PD进展的治疗方法。