Department of Pharmacology and Physiology, Université de Montréal, Institute for Research in Immunology and Cancer, QC, Canada.
Department of Translational Medicine, School of Medical Sciences, University of Campinas, Campinas, Brazil.
Front Immunol. 2022 Mar 24;13:867443. doi: 10.3389/fimmu.2022.867443. eCollection 2022.
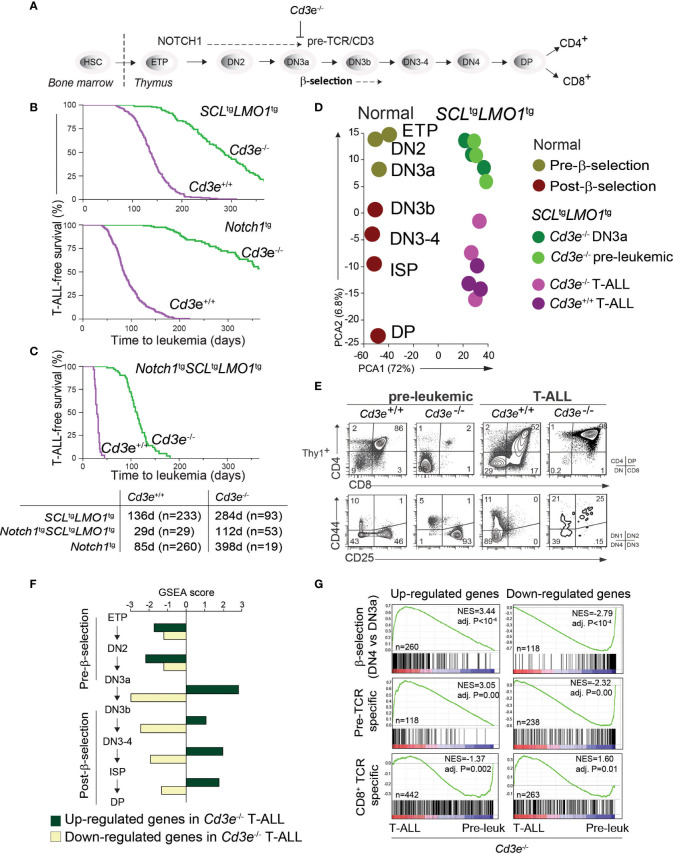
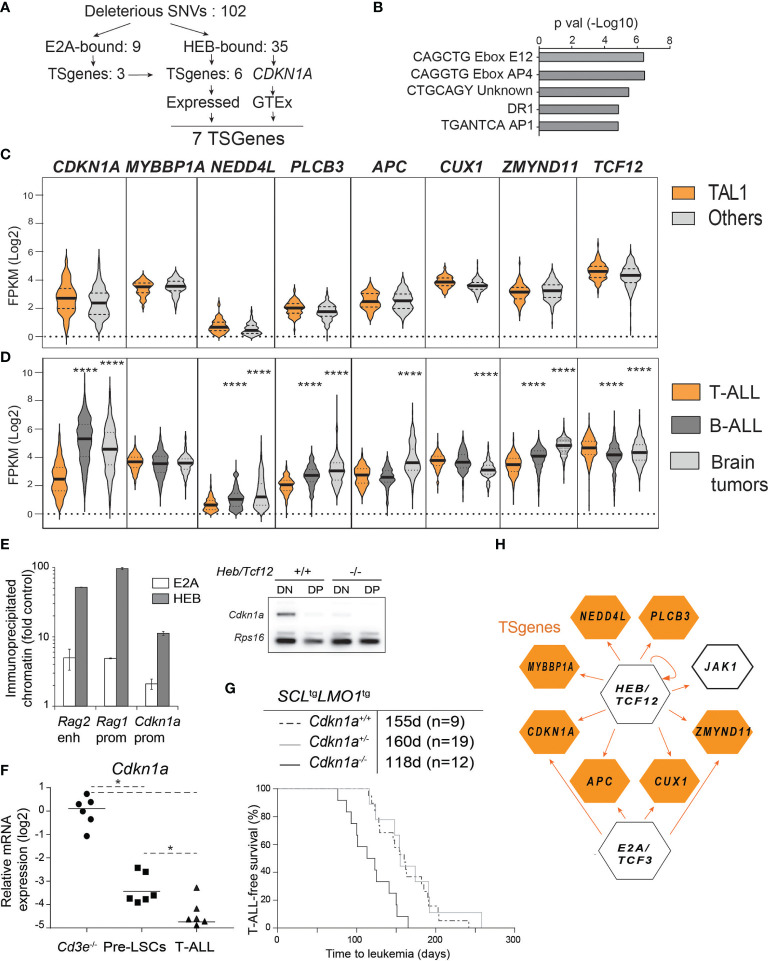
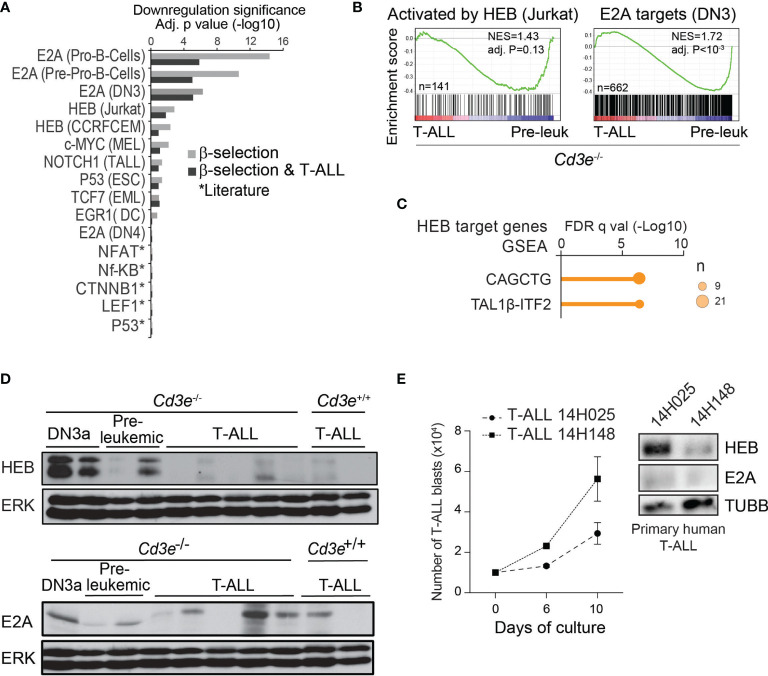
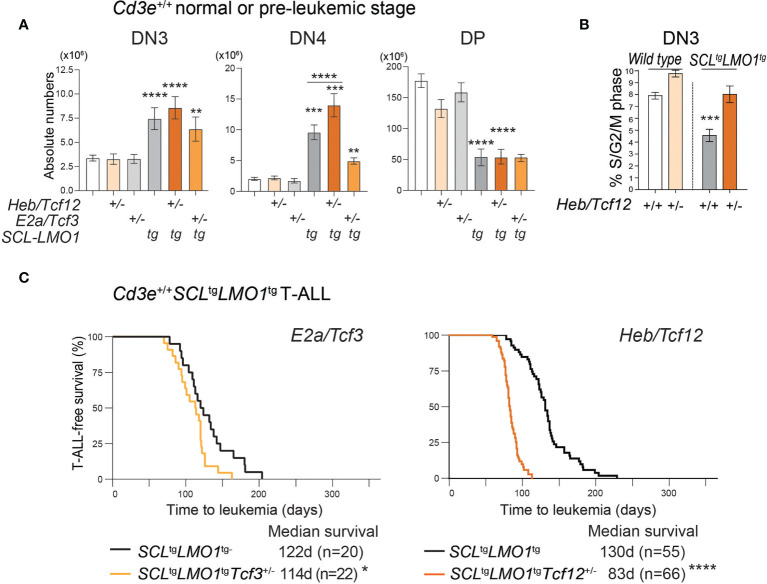
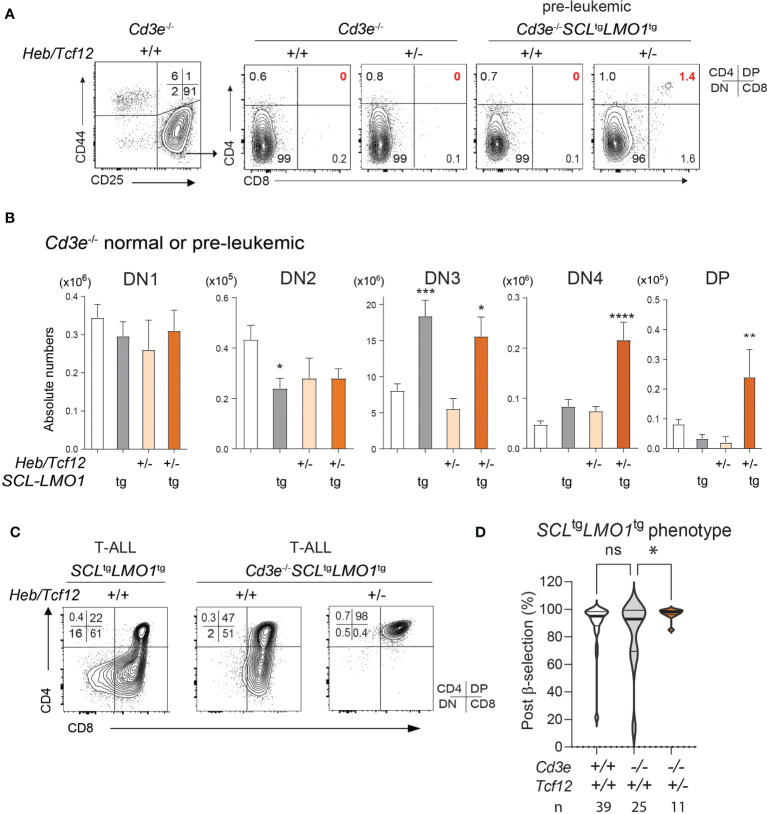
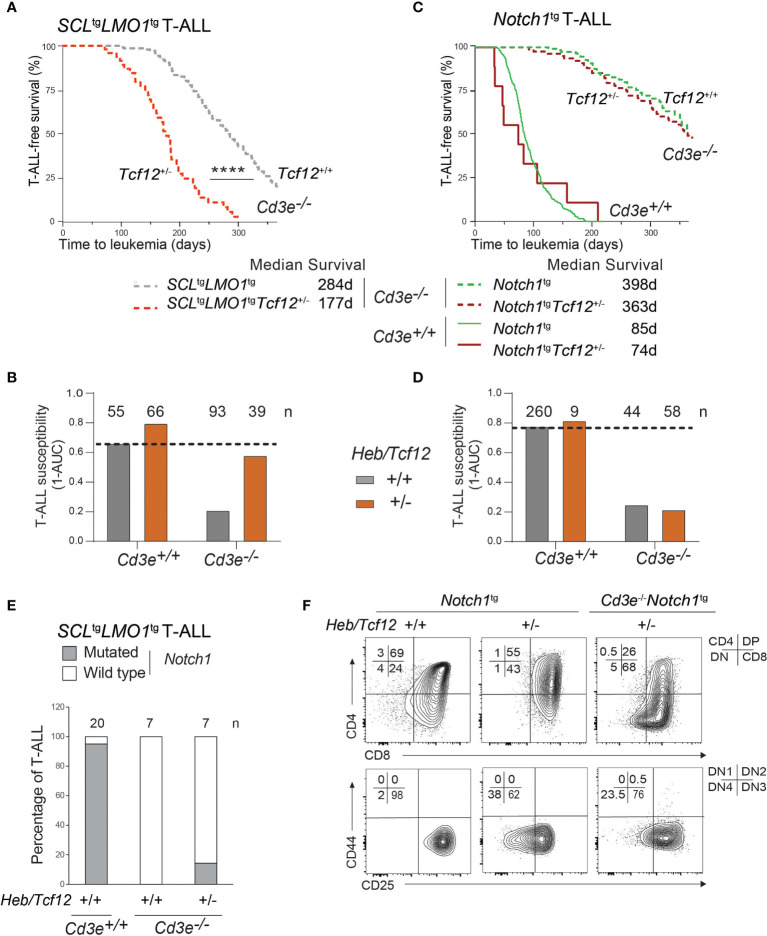
Early T-cell development is precisely controlled by E proteins, that indistinguishably include HEB/TCF12 and E2A/TCF3 transcription factors, together with NOTCH1 and pre-T cell receptor (TCR) signalling. Importantly, perturbations of early T-cell regulatory networks are implicated in leukemogenesis. NOTCH1 gain of function mutations invariably lead to T-cell acute lymphoblastic leukemia (T-ALL), whereas inhibition of E proteins accelerates leukemogenesis. Thus, NOTCH1, pre-TCR, E2A and HEB functions are intertwined, but how these pathways contribute individually or synergistically to leukemogenesis remain to be documented. To directly address these questions, we leveraged -deficient mice in which pre-TCR signaling and progression through β-selection is abrogated to dissect and decouple the roles of pre-TCR, NOTCH1, E2A and HEB in SCL/TAL1-induced T-ALL, the use of gain of function transgenic () and or heterozygote mice. As a result, we now provide evidence that both HEB and E2A restrain cell proliferation at the β-selection checkpoint while the clonal expansion of SCL-LMO1-induced pre-leukemic stem cells in T-ALL is uniquely dependent on gene dosage. At the molecular level, HEB protein levels are decreased proteasomal degradation at the leukemic stage, pointing to a reversible loss of function mechanism. Moreover, in -induced T-ALL, loss of one allele is sufficient to bypass pre-TCR signaling which is required for gain of function mutations and for progression to T-ALL. In contrast, monoallelic deletion does not accelerate -induced T-ALL, indicating that and operate in the same pathway. Finally, we identify a tumor suppressor gene set downstream of HEB, exhibiting significantly lower expression levels in pediatric T-ALL compared to B-ALL and brain cancer samples, the three most frequent pediatric cancers. In summary, our results indicate a tumor suppressor function of HEB/TCF12 in T-ALL to mitigate cell proliferation controlled by NOTCH1 in pre-leukemic stem cells and prevent NOTCH1-driven progression to T-ALL.
早期 T 细胞的发育受到 E 蛋白的精确调控,E 蛋白包括 HEB/TCF12 和 E2A/TCF3 转录因子,以及 NOTCH1 和前 T 细胞受体 (TCR) 信号通路。重要的是,早期 T 细胞调控网络的扰动与白血病的发生有关。NOTCH1 功能获得性突变总是导致 T 细胞急性淋巴细胞白血病 (T-ALL),而 E 蛋白的抑制会加速白血病的发生。因此,NOTCH1、pre-TCR、E2A 和 HEB 的功能相互交织,但这些途径如何单独或协同促进白血病的发生仍有待记录。为了直接解决这些问题,我们利用 pre-TCR 信号缺失的 -/- 小鼠,这些小鼠的β选择过程被阻断,以剖析和分离 pre-TCR、NOTCH1、E2A 和 HEB 在 SCL/TAL1 诱导的 T-ALL 中的作用,使用 gain of function 转基因 () 和 或 杂合子小鼠。结果表明,HEB 和 E2A 都在β选择检查点上抑制细胞增殖,而 SCL-LMO1 诱导的前白血病干细胞在 T-ALL 中的克隆扩增则完全依赖于 基因剂量。在分子水平上,HEB 蛋白水平在白血病阶段通过蛋白酶体降解而降低,这表明存在可逆的功能丧失机制。此外,在 -/- 诱导的 T-ALL 中,缺失一个 等位基因足以绕过 pre-TCR 信号,该信号对于 功能获得性突变和向 T-ALL 的进展是必需的。相比之下,单等位基因缺失不会加速 -/- 诱导的 T-ALL,表明 和 作用于相同的途径。最后,我们鉴定了一组下游的抑癌基因,与 B-ALL 和脑癌样本相比,这些基因在儿科 T-ALL 中的表达水平明显较低,这三种是最常见的儿科癌症。总之,我们的结果表明,HEB/TCF12 在 T-ALL 中具有肿瘤抑制功能,可减轻 pre-leukemic 干细胞中 NOTCH1 控制的细胞增殖,并阻止 NOTCH1 驱动的向 T-ALL 的进展。