Lin Shaofeng, Chen Yanping, Wang Jianchao, Cai Yibin, Chen Xiaohui, Chen Yuanmei, Shi Yi, Chen Gang, Zhu Kunshou
Department of Thoracic Surgery, Fujian Medical University Cancer Hospital and Fujian Cancer Hospital, Fuzhou, China.
Department of Pathology, Fujian Medical University Cancer Hospital and Fujian Cancer Hospital, Fuzhou, China.
Front Genet. 2022 Mar 23;13:830601. doi: 10.3389/fgene.2022.830601. eCollection 2022.
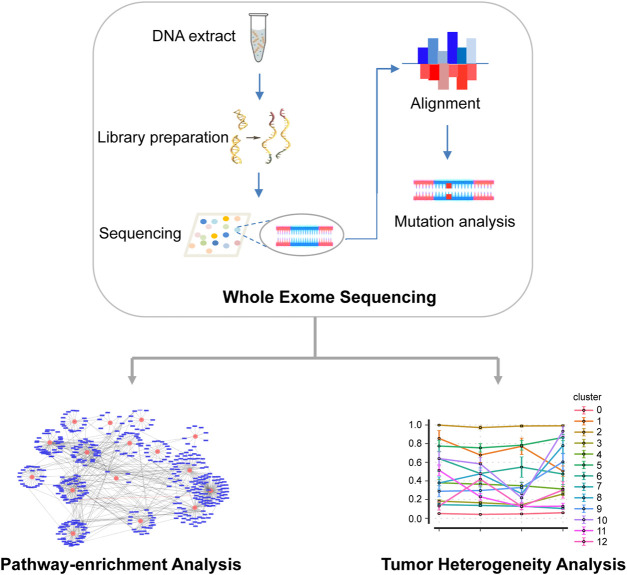
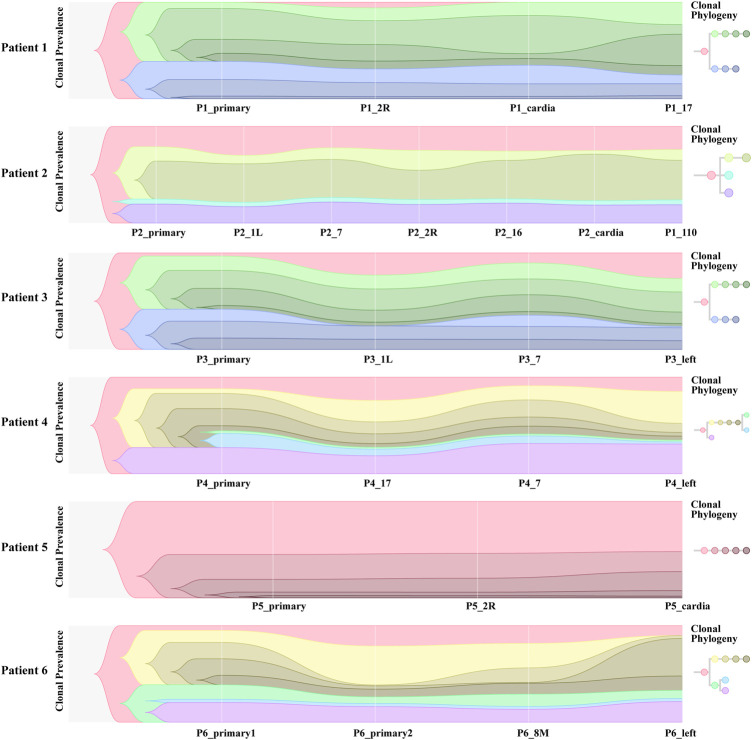
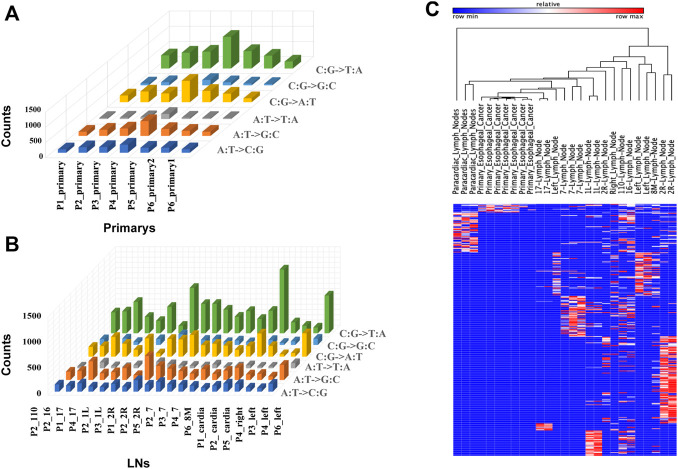
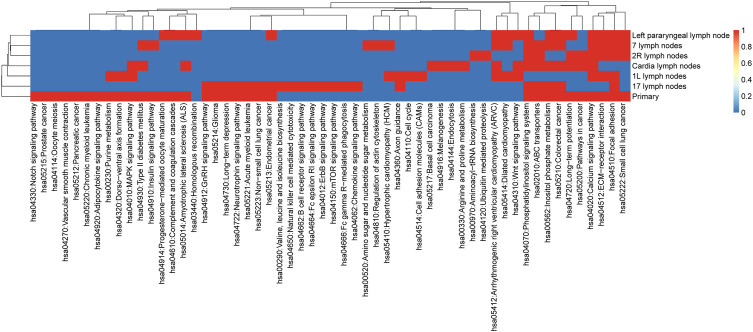
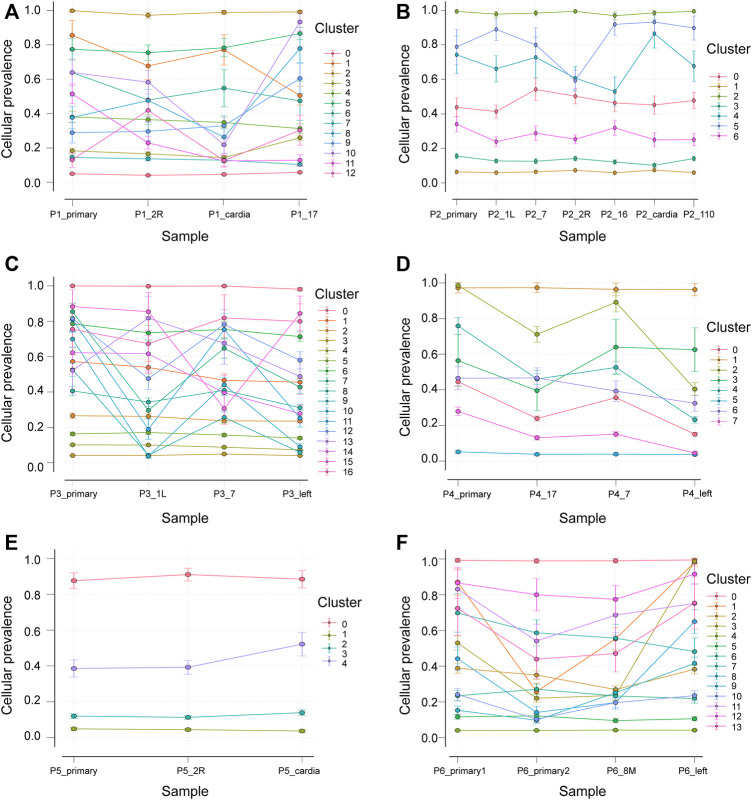
Esophageal cancer is an aggressive malignant tumor, with 90 percent of the patients prone to recurrence and metastasis. Although recent studies have identified some potential biomarkers, these biomarkers' clinical or pathological significance is still unclear. Therefore, it is urgent to further identify and study novel molecular changes occurring in esophageal cancer. It has positive clinical significance to identify a tumor-specific mutation in patients after surgery for an effective intervention to improve the prognosis of patients. In this study, we performed whole-exome sequencing (WES) on 33 tissue samples from six esophageal cancer patients with lymph node metastasis, compared the differences in the genomic and evolutionary maps in different tissues, and then performed pathway enrichment analysis on non-synonymous mutation genes. Finally, we sorted out the somatic mutation data of all patients to analyze the subclonality of each tumor. There were significant differences in somatic mutations between the metastatic lymph nodes and primary lesions in the six patients. Clustering results of pathway enrichment analysis indicated that the metastatic lymph nodes had certain commonalities. Tumors of the cloned exploration results illustrated that five patients showed substantial heterogeneity. WES technology can be used to explore the differences in regional evolutionary maps, heterogeneity, and detect patients' tumor-specific mutations. In addition, an in-depth understanding of the ontogeny and phylogeny of tumor heterogeneity can help to further find new molecular changes in esophageal cancer, which can improve the prognosis of EC patients and provide a valuable reference for their diagnosis.
食管癌是一种侵袭性恶性肿瘤,90%的患者容易复发和转移。尽管最近的研究已经确定了一些潜在的生物标志物,但这些生物标志物的临床或病理意义仍不清楚。因此,迫切需要进一步识别和研究食管癌中发生的新的分子变化。识别患者术后的肿瘤特异性突变以便进行有效干预以改善患者预后具有积极的临床意义。在本研究中,我们对6例伴有淋巴结转移的食管癌患者的33个组织样本进行了全外显子组测序(WES),比较了不同组织中基因组和进化图谱的差异,然后对非同义突变基因进行了通路富集分析。最后,我们整理了所有患者的体细胞突变数据以分析每个肿瘤的亚克隆性。6例患者的转移淋巴结和原发灶之间的体细胞突变存在显著差异。通路富集分析的聚类结果表明转移淋巴结有一定的共性。克隆探索结果的肿瘤显示5例患者表现出显著的异质性。WES技术可用于探索区域进化图谱的差异、异质性,并检测患者的肿瘤特异性突变。此外,深入了解肿瘤异质性的个体发生和系统发育有助于进一步发现食管癌中的新分子变化,这可以改善食管癌患者的预后并为其诊断提供有价值的参考。