Department of Quantitative Health Sciences, Center for Individualized Medicine, Mayo Clinic, Rochester, Minnesota, United States of America.
Child Study Center, Yale University, New Haven, Connecticut, United States of America.
PLoS Comput Biol. 2022 Apr 20;18(4):e1009487. doi: 10.1371/journal.pcbi.1009487. eCollection 2022 Apr.
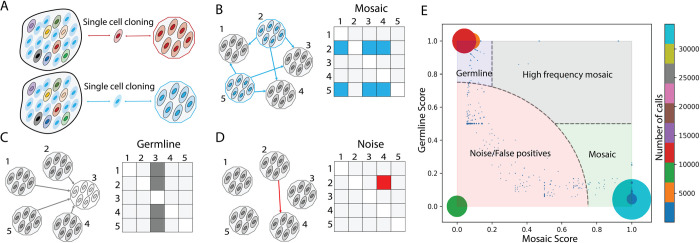
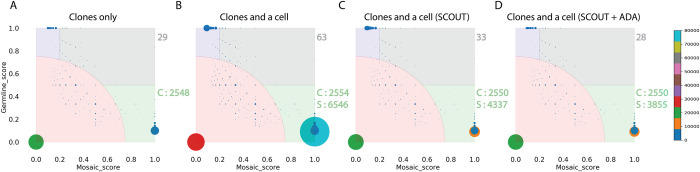
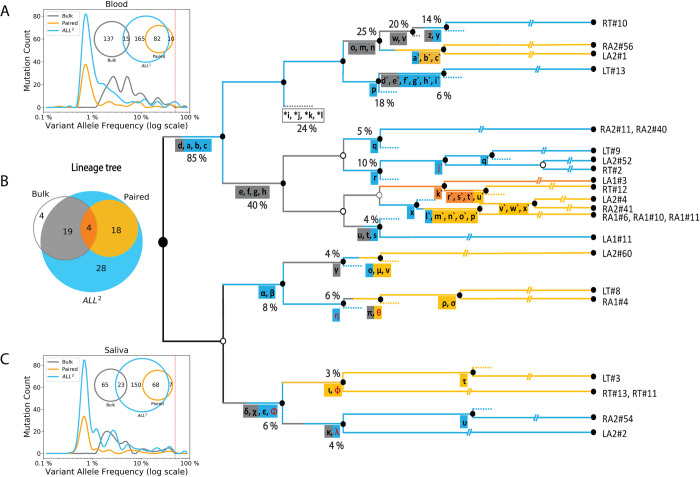
Accurate discovery of somatic mutations in a cell is a challenge that partially lays in immaturity of dedicated analytical approaches. Approaches comparing a cell's genome to a control bulk sample miss common mutations, while approaches to find such mutations from bulk suffer from low sensitivity. We developed a tool, All2, which enables accurate filtering of mutations in a cell without the need for data from bulk(s). It is based on pair-wise comparisons of all cells to each other where every call for base pair substitution and indel is classified as either a germline variant, mosaic mutation, or false positive. As All2 allows for considering dropped-out regions, it is applicable to whole genome and exome analysis of cloned and amplified cells. By applying the approach to a variety of available data, we showed that its application reduces false positives, enables sensitive discovery of high frequency mutations, and is indispensable for conducting high resolution cell lineage tracing.
准确发现细胞中的体细胞突变是一个挑战,部分原因在于专门的分析方法不够成熟。将细胞的基因组与对照批量样本进行比较的方法会遗漏常见的突变,而从批量样本中寻找此类突变的方法则存在灵敏度低的问题。我们开发了一种名为 All2 的工具,它无需批量数据即可实现对细胞中突变的准确筛选。它基于对所有细胞进行两两比较,其中每个碱基替换和插入缺失的调用都被分类为种系变异、镶嵌突变或假阳性。由于 All2 允许考虑掉失区域,因此它适用于克隆和扩增细胞的全基因组和外显子分析。通过将该方法应用于各种可用数据,我们表明它的应用可以减少假阳性,能够敏感地发现高频突变,并且对于进行高分辨率细胞谱系追踪是不可或缺的。