German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany.
InVivoBiosystems, 1505 Westec Dr, Eugene, OR, USA.
Mol Metab. 2022 Jul;61:101503. doi: 10.1016/j.molmet.2022.101503. Epub 2022 Apr 19.
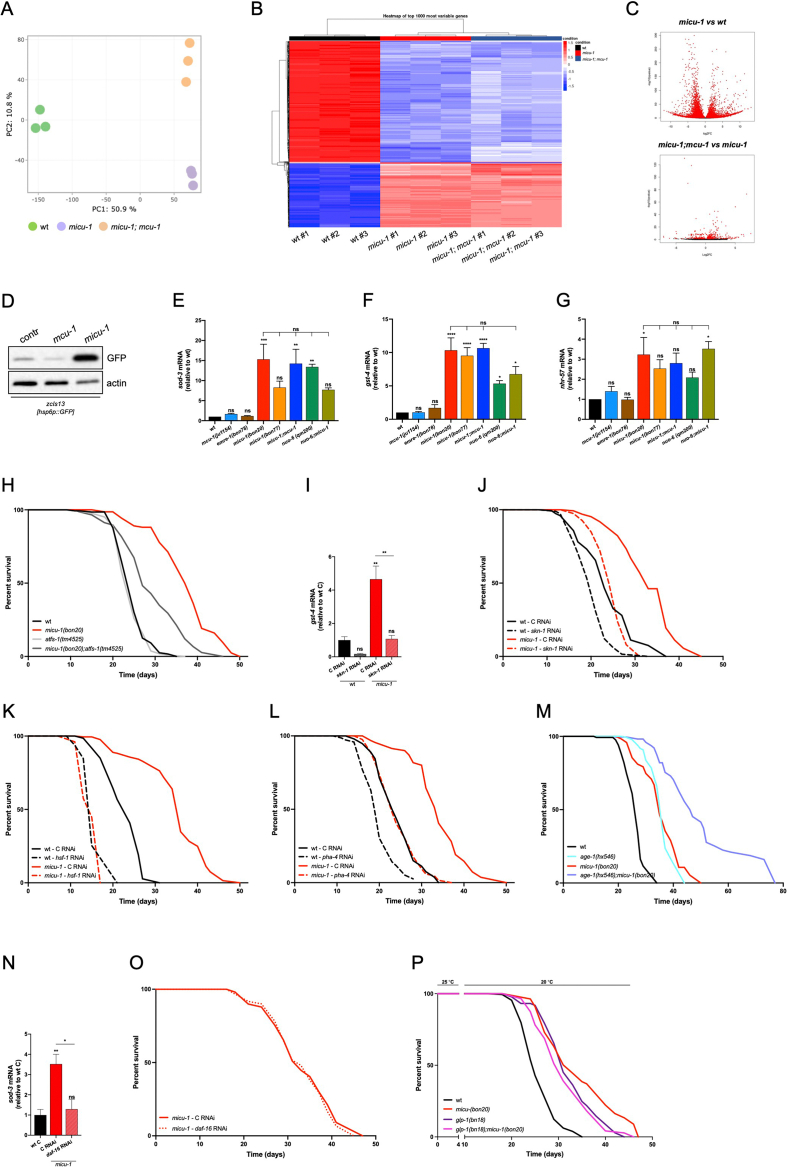
Mitochondrial "retrograde" signaling may stimulate organelle biogenesis as a compensatory adaptation to aberrant activity of the oxidative phosphorylation (OXPHOS) system. To maintain energy-consuming processes in OXPHOS deficient cells, alternative metabolic pathways are functionally coupled to the degradation, recycling and redistribution of biomolecules across distinct intracellular compartments. While transcriptional regulation of mitochondrial network expansion has been the focus of many studies, the molecular mechanisms promoting mitochondrial maintenance in energy-deprived cells remain poorly investigated.
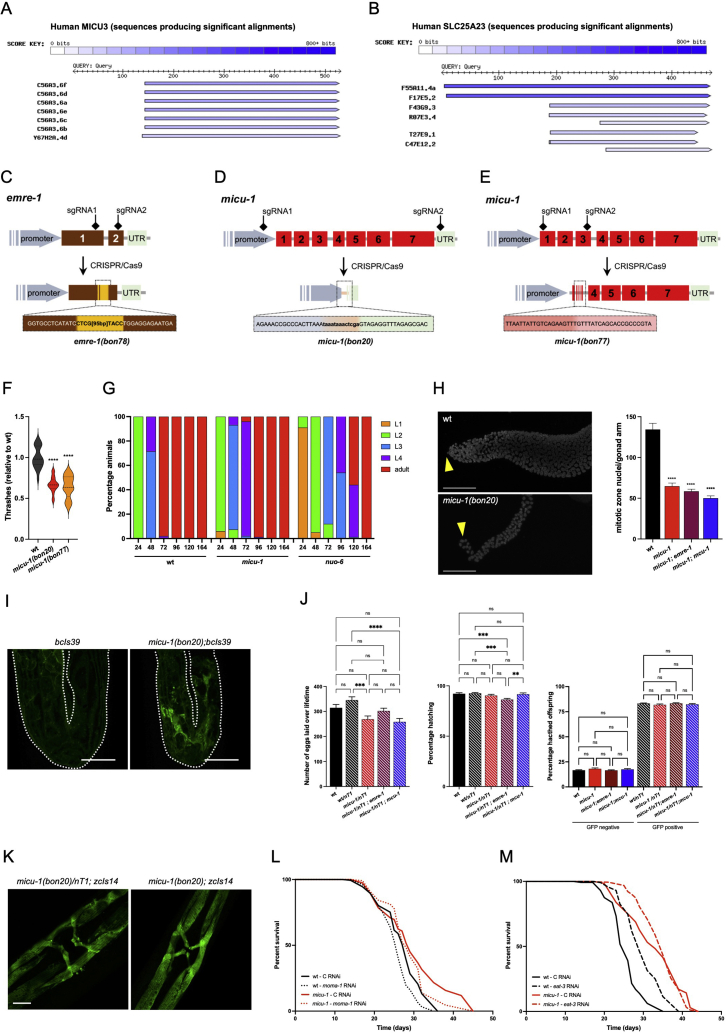
We performed transcriptomics, quantitative proteomics and lifespan assays to identify pathways that are mechanistically linked to mitochondrial network expansion and homeostasis in Caenorhabditis elegans lacking the mitochondrial calcium uptake protein 1 (MICU-1/MICU1). To support our findings, we carried out biochemical and image analyses in mammalian cells and mouse-derived tissues.
We report that micu-1(null) mutations impair the OXPHOS system and promote C. elegans longevity through a transcriptional program that is independent of the mitochondrial calcium uniporter MCU-1/MCU and the essential MCU regulator EMRE-1/EMRE. We identify sphingosine phosphate lyase SPL-1/SGPL1 and the ATFS-1-target HOPS complex subunit VPS-39/VPS39 as critical lifespan modulators of micu-1(null) mutant animals. Cross-species investigation indicates that SGPL1 upregulation stimulates VPS39 recruitment to the mitochondria, thereby enhancing mitochondria-lysosome contacts. Consistently, VPS39 downregulation compromises mitochondrial network maintenance and basal autophagic flux in MICU1 deficient cells. In mouse-derived muscles, we show that VPS39 recruitment to the mitochondria may represent a common signature associated with altered OXPHOS system.
Our findings reveal a previously unrecognized SGPL1/VPS39 axis that stimulates intracellular organelle interactions and sustains autophagy and mitochondrial homeostasis in OXPHOS deficient cells.
线粒体“逆行”信号可能会刺激细胞器生物发生,作为氧化磷酸化(OXPHOS)系统异常活性的代偿适应。为了维持 OXPHOS 缺陷细胞中耗能过程,替代代谢途径在功能上与生物分子在不同细胞内隔室中的降解、回收和再分配耦联。虽然线粒体网络扩张的转录调控一直是许多研究的焦点,但在能量匮乏的细胞中促进线粒体维持的分子机制仍未得到充分研究。
我们进行了转录组学、定量蛋白质组学和寿命测定,以确定与线粒体网络扩张和 Caenorhabditis elegans 中线粒体钙摄取蛋白 1(MICU-1/MICU1)缺失中线粒体稳态相关的机制途径。为了支持我们的发现,我们在哺乳动物细胞和鼠源性组织中进行了生化和图像分析。
我们报告说,micu-1(null)突变会损害 OXPHOS 系统,并通过一种独立于线粒体钙单向转运蛋白 MCU-1/MCU 和必需的 MCU 调节剂 EMRE-1/EMRE 的转录程序促进 C. elegans 的长寿。我们确定了神经酰胺磷酸酶 SPL-1/SGPL1 和 ATFS-1 靶标 HOPS 复合物亚基 VPS-39/VPS39 作为 micu-1(null)突变体动物的关键寿命调节剂。跨物种研究表明,SGPL1 的上调刺激了 VPS39 向线粒体的募集,从而增强了线粒体-溶酶体接触。一致地,VPS39 的下调会损害 MICU1 缺陷细胞中线粒体网络的维持和基础自噬通量。在鼠源性肌肉中,我们表明 VPS39 向线粒体的募集可能代表与改变的 OXPHOS 系统相关的共同特征。
我们的研究结果揭示了一个以前未被识别的 SGPL1/VPS39 轴,该轴刺激细胞内细胞器相互作用,并维持 OXPHOS 缺陷细胞中的自噬和线粒体稳态。