Centre for Personalised Immunology, Department of Immunology and Infectious Disease, John Curtin School of Medical Research, Australian National University, Canberra, Australian Capital Territory, Australia.
Research School of Biology, Australian National University, Canberra, Australian Capital Territory, Australia.
Nature. 2022 May;605(7909):349-356. doi: 10.1038/s41586-022-04642-z. Epub 2022 Apr 27.
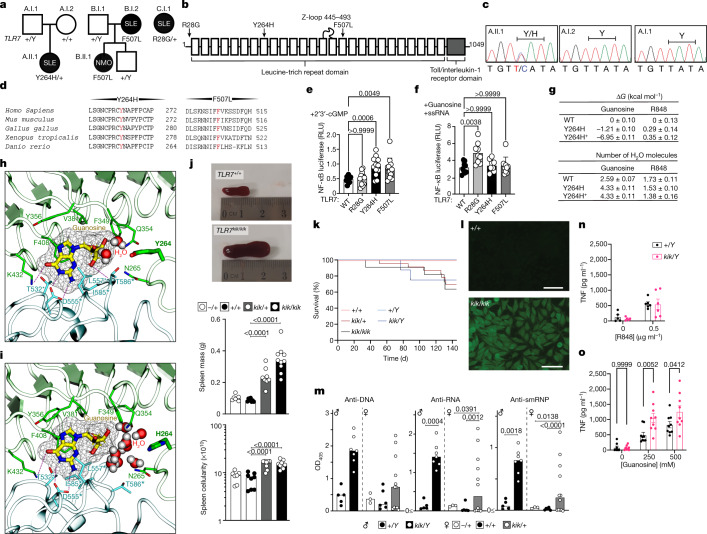
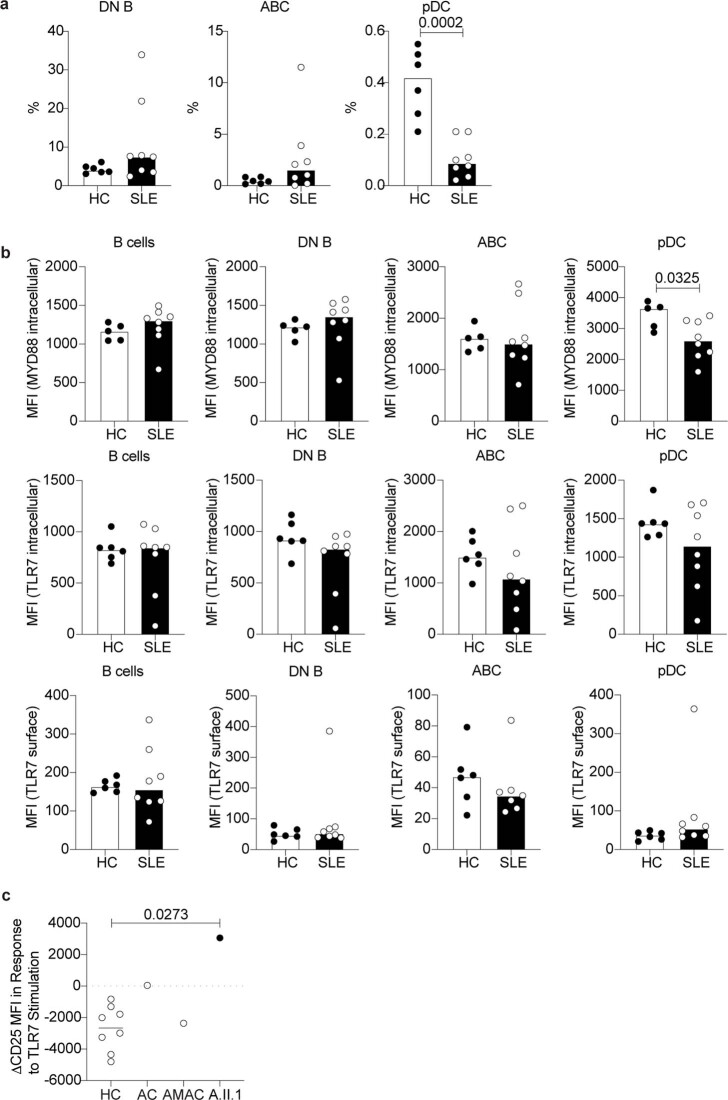
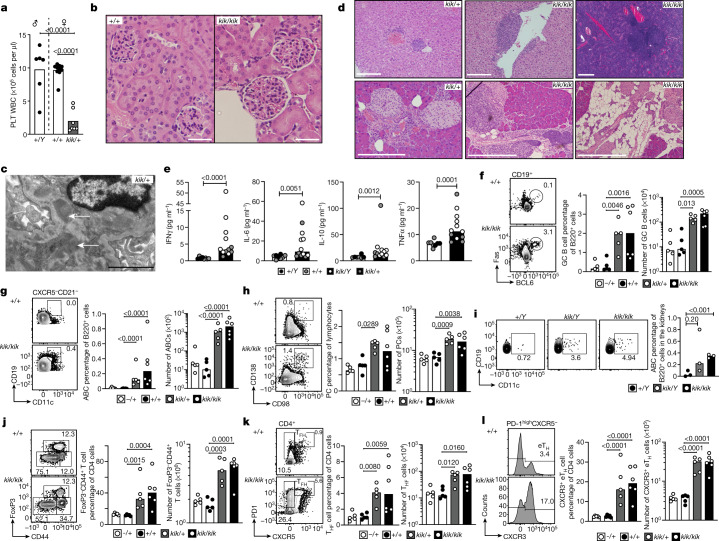
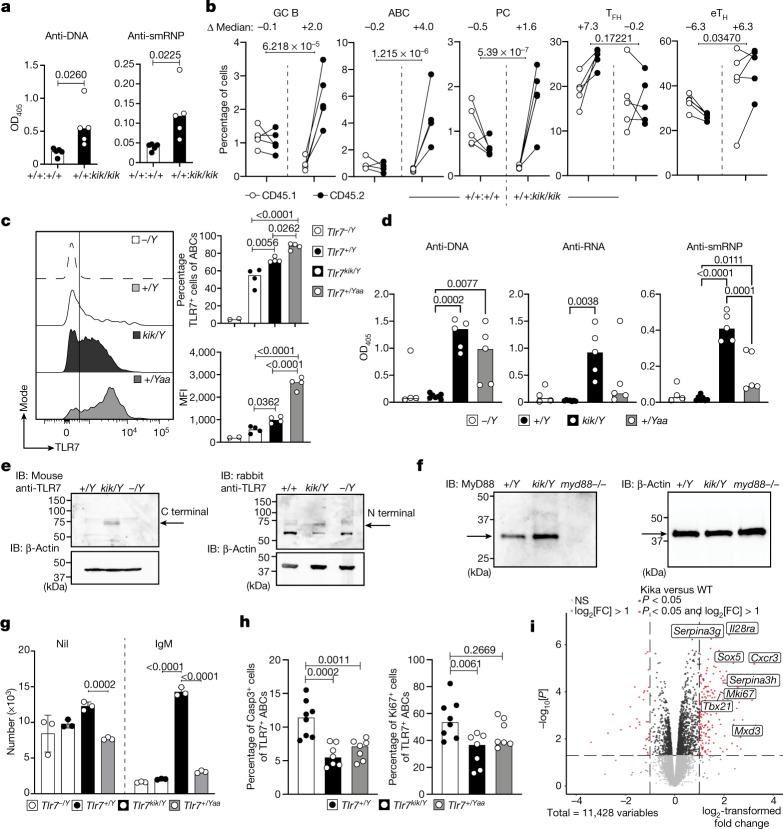
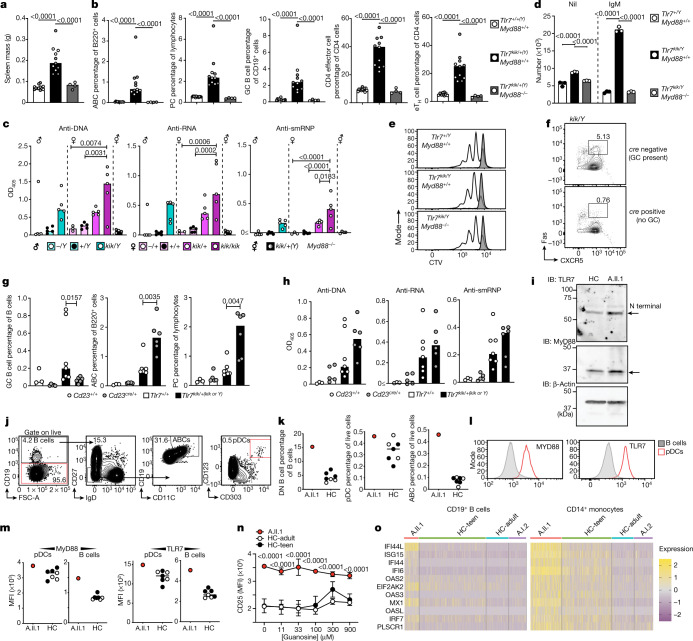
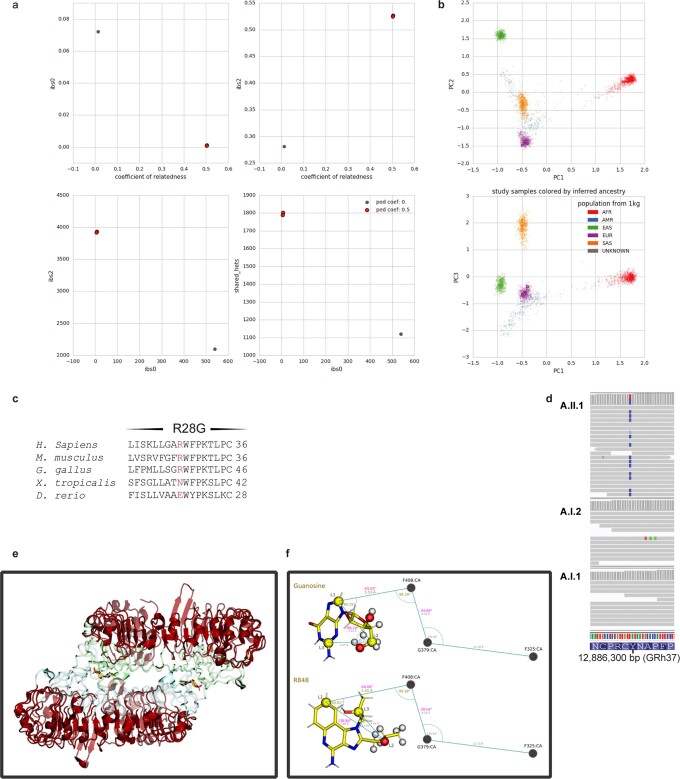
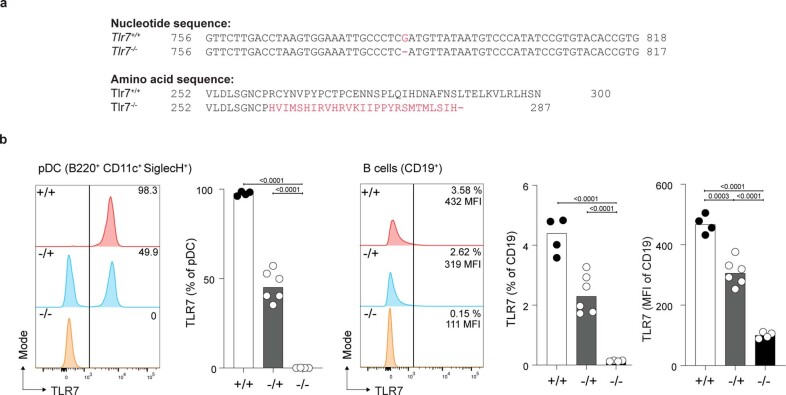
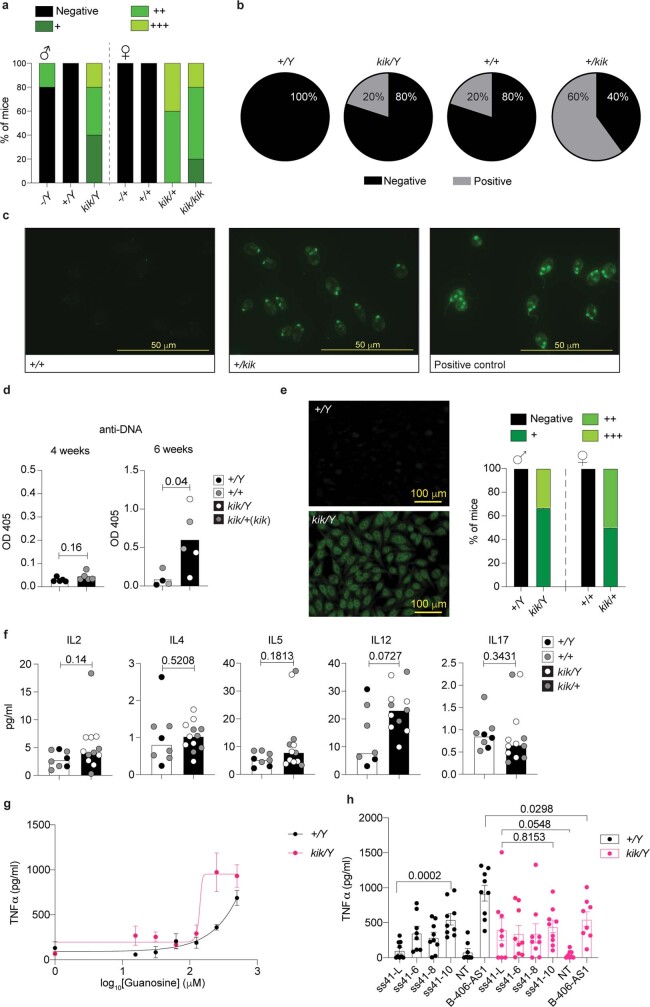
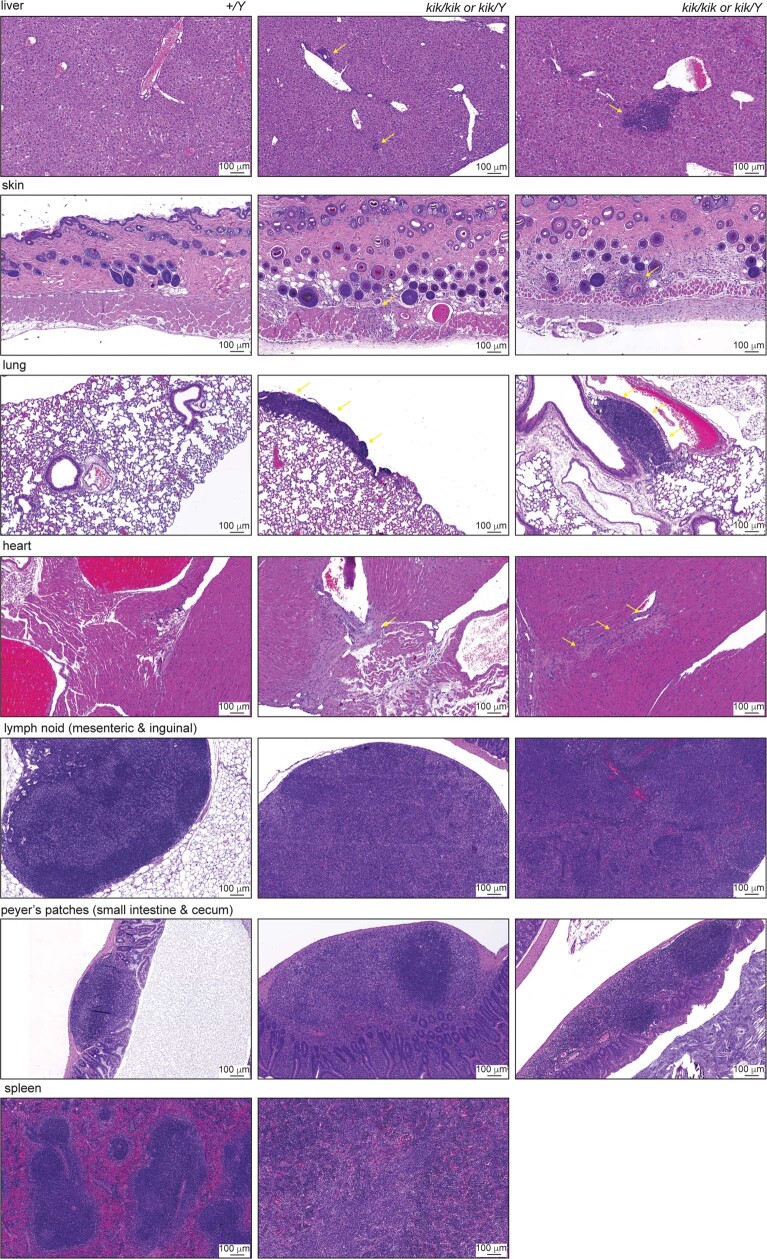
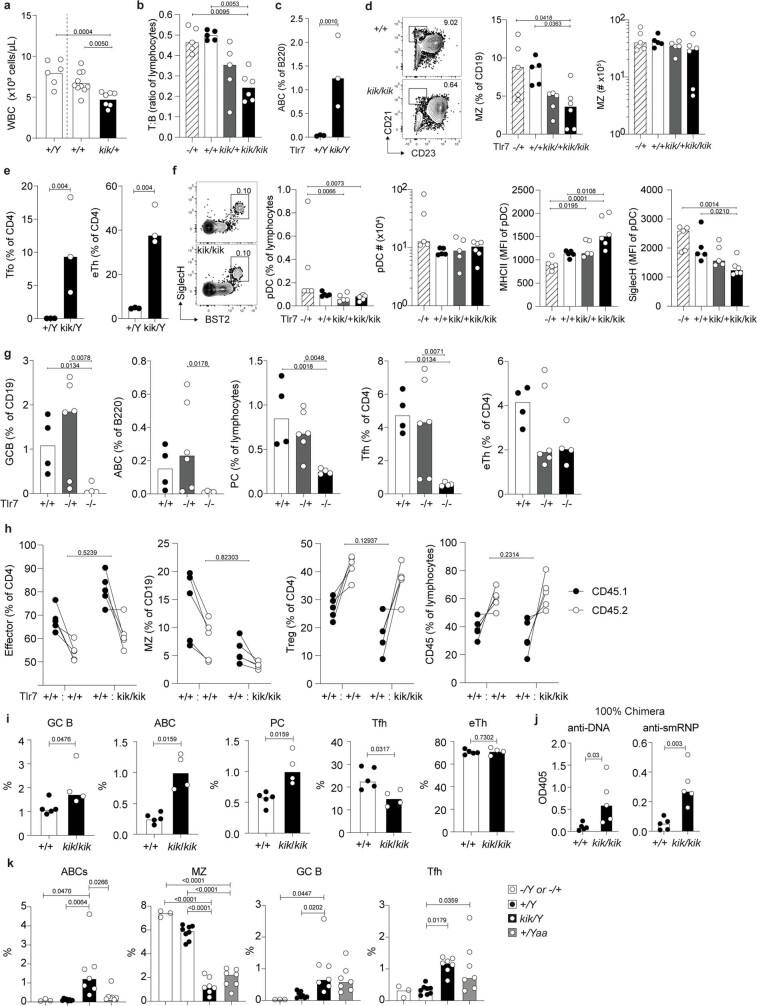
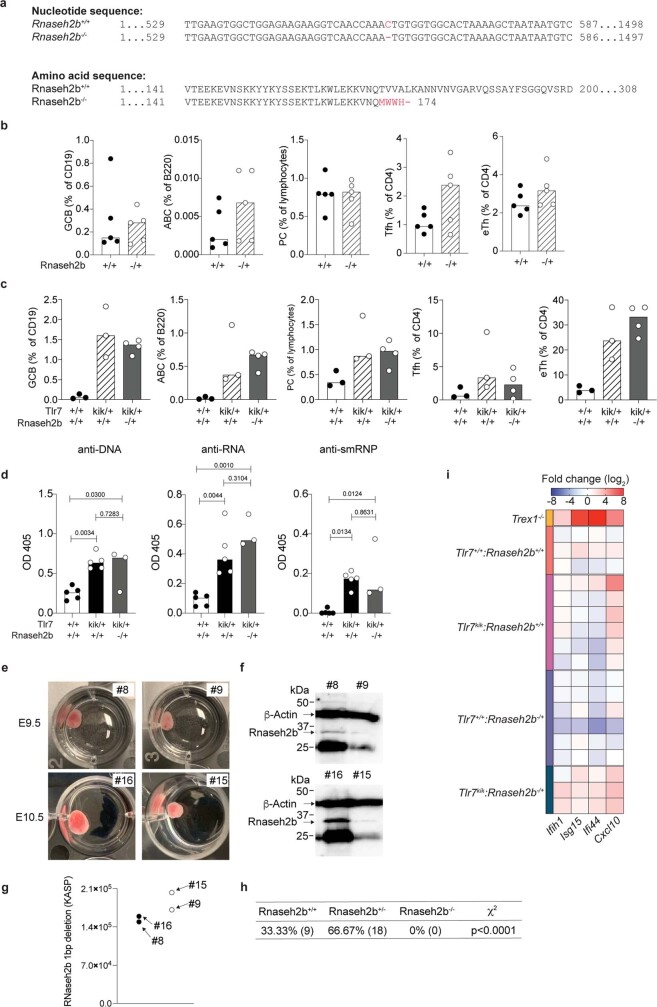
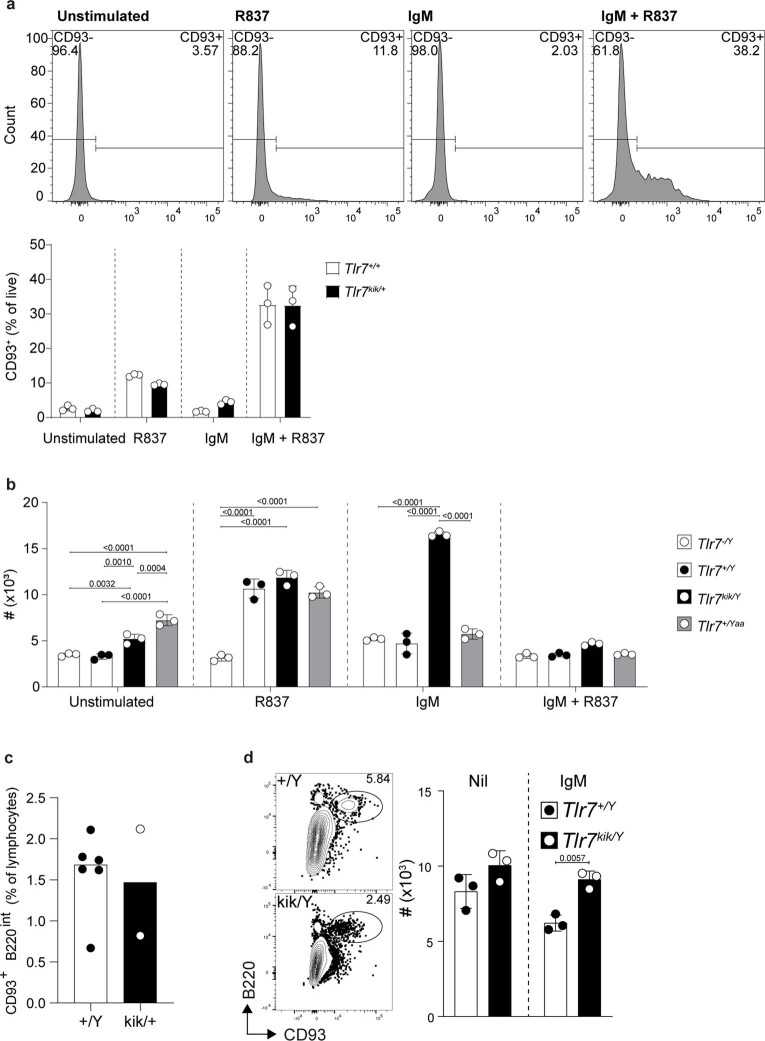
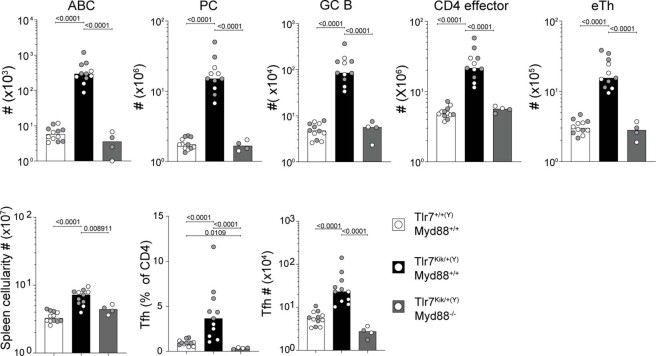
Although circumstantial evidence supports enhanced Toll-like receptor 7 (TLR7) signalling as a mechanism of human systemic autoimmune disease, evidence of lupus-causing TLR7 gene variants is lacking. Here we describe human systemic lupus erythematosus caused by a TLR7 gain-of-function variant. TLR7 is a sensor of viral RNA, and binds to guanosine-. We identified a de novo, previously undescribed missense TLR7 variant in a child with severe lupus and additional variants in other patients with lupus. The TLR7 variant selectively increased sensing of guanosine and 2',3'-cGMP, and was sufficient to cause lupus when introduced into mice. We show that enhanced TLR7 signalling drives aberrant survival of B cell receptor (BCR)-activated B cells, and in a cell-intrinsic manner, accumulation of CD11c age-associated B cells and germinal centre B cells. Follicular and extrafollicular helper T cells were also increased but these phenotypes were cell-extrinsic. Deficiency of MyD88 (an adaptor protein downstream of TLR7) rescued autoimmunity, aberrant B cell survival, and all cellular and serological phenotypes. Despite prominent spontaneous germinal-centre formation in Tlr7 mice, autoimmunity was not ameliorated by germinal-centre deficiency, suggesting an extrafollicular origin of pathogenic B cells. We establish the importance of TLR7 and guanosine-containing self-ligands for human lupus pathogenesis, which paves the way for therapeutic TLR7 or MyD88 inhibition.
虽然间接证据支持 Toll 样受体 7(TLR7)信号增强是人类全身性自身免疫性疾病的一种机制,但缺乏导致狼疮的 TLR7 基因突变的证据。在这里,我们描述了一种由 TLR7 功能获得性突变引起的人类系统性红斑狼疮。TLR7 是病毒 RNA 的传感器,与鸟嘌呤核苷酸结合。我们在一名患有严重狼疮的儿童中发现了一个新的、以前未描述的 TLR7 错义突变,以及其他狼疮患者中的其他变体。TLR7 变体选择性地增加了对鸟嘌呤和 2',3'-cGMP 的感知,并且当引入小鼠时足以引起狼疮。我们表明,增强的 TLR7 信号传导导致 B 细胞受体(BCR)激活的 B 细胞的异常存活,并且以细胞内固有方式,导致 CD11c 年龄相关 B 细胞和生发中心 B 细胞的积累。滤泡性和滤泡外辅助性 T 细胞也增加了,但这些表型是细胞外在的。MyD88(TLR7 下游的衔接蛋白)的缺失挽救了自身免疫、异常的 B 细胞存活以及所有细胞和血清学表型。尽管 Tlr7 小鼠中自发生发中心形成明显,但生发中心缺陷并不能改善自身免疫,这表明致病性 B 细胞的起源是滤泡外的。我们确定了 TLR7 和含有鸟嘌呤的自身配体对人类狼疮发病机制的重要性,这为治疗性 TLR7 或 MyD88 抑制铺平了道路。