Department of Immunobiology, Yale University School of Medicine, New Haven, CT, USA.
Department of Pathology, Yale University School of Medicine, New Haven, CT, USA.
Nature. 2022 Jun;606(7914):585-593. doi: 10.1038/s41586-022-04802-1. Epub 2022 Apr 28.
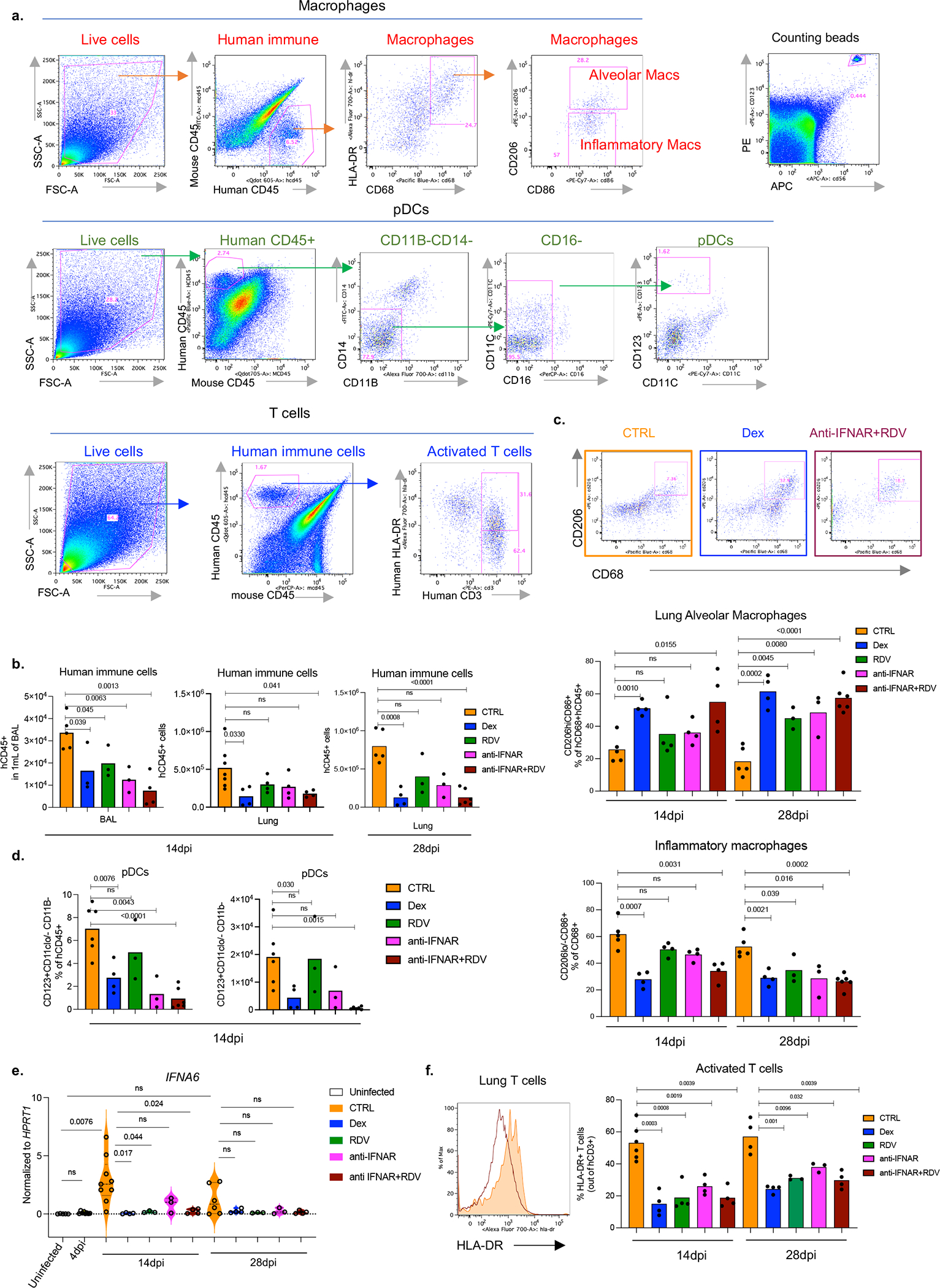
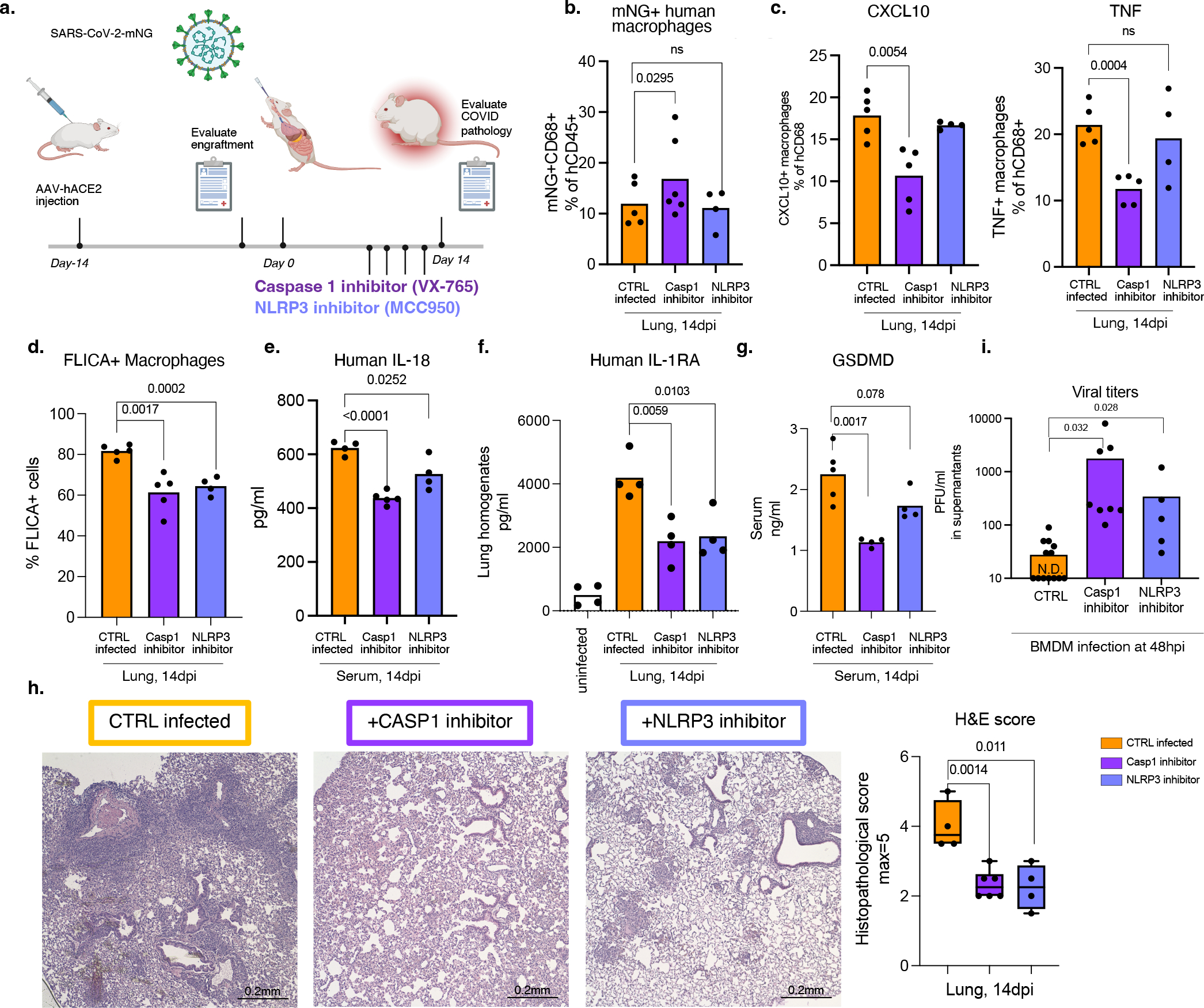
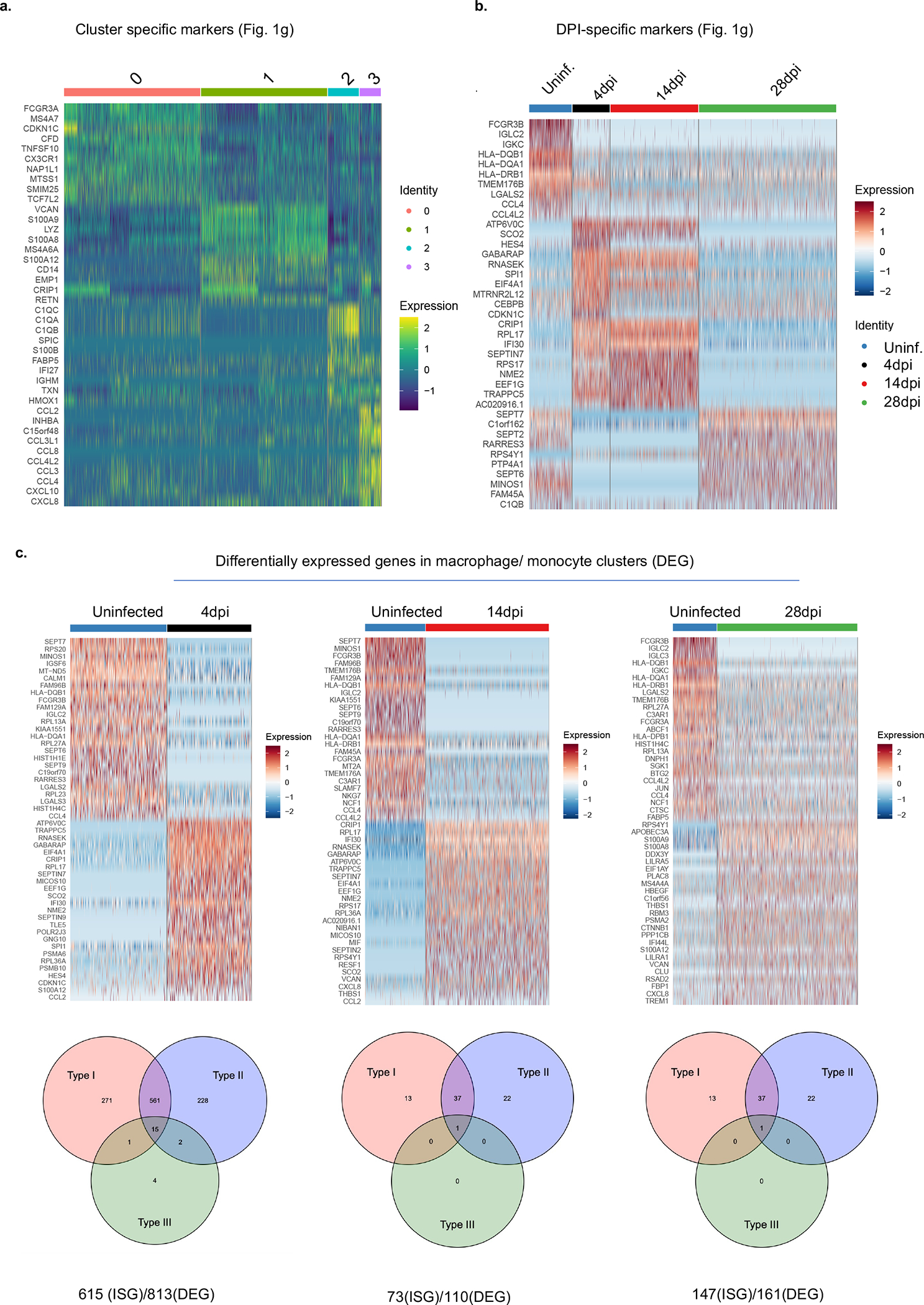
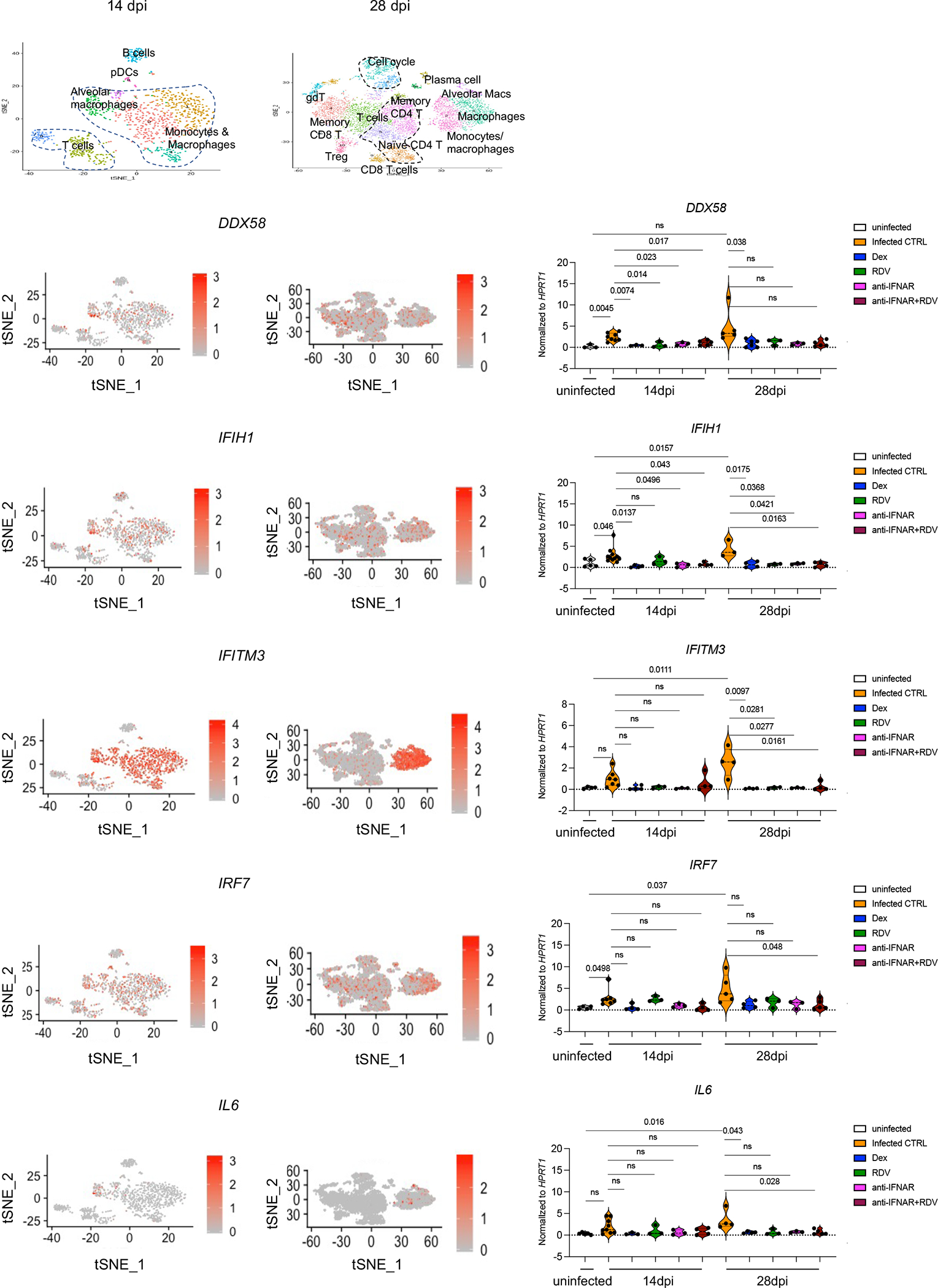
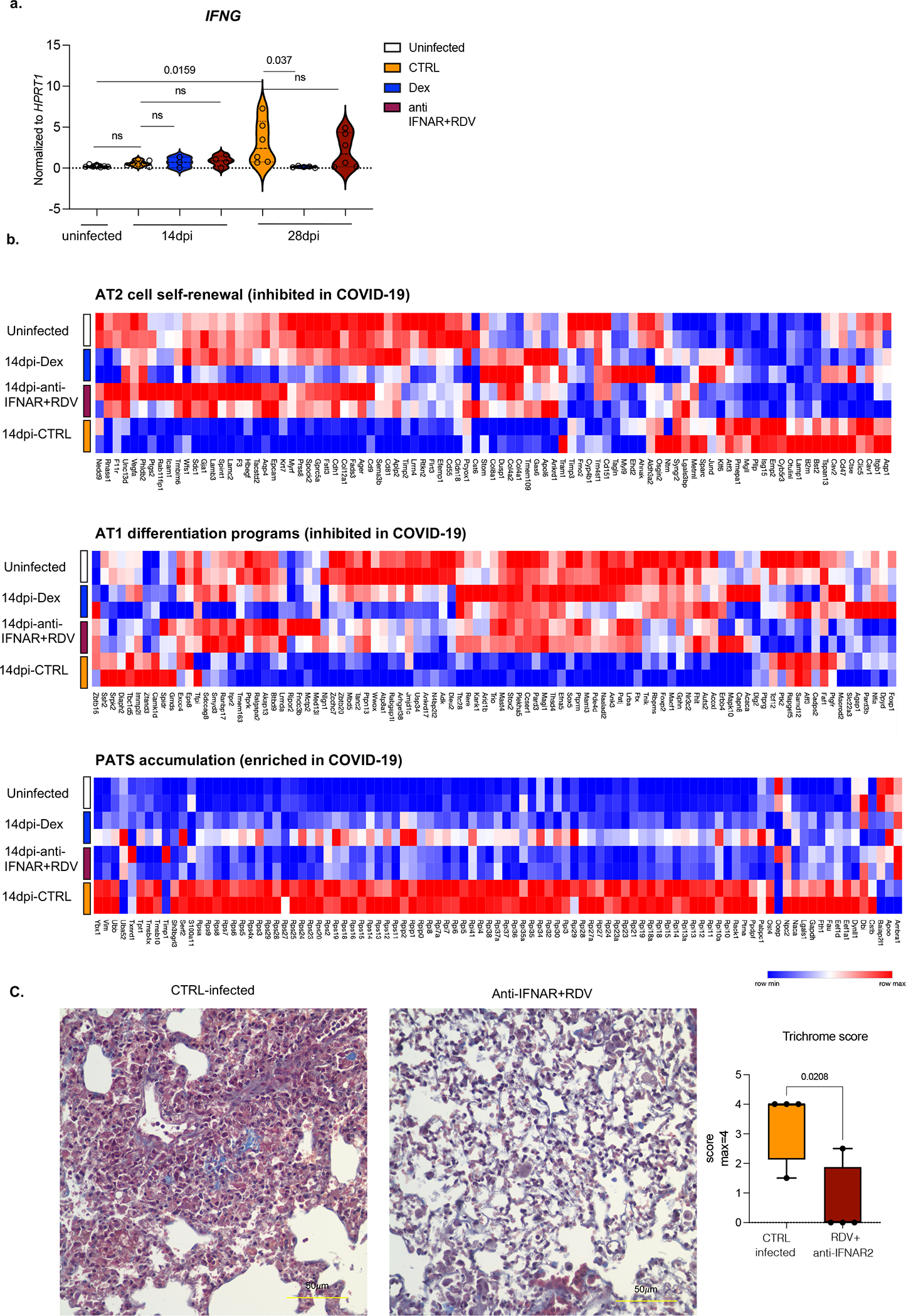
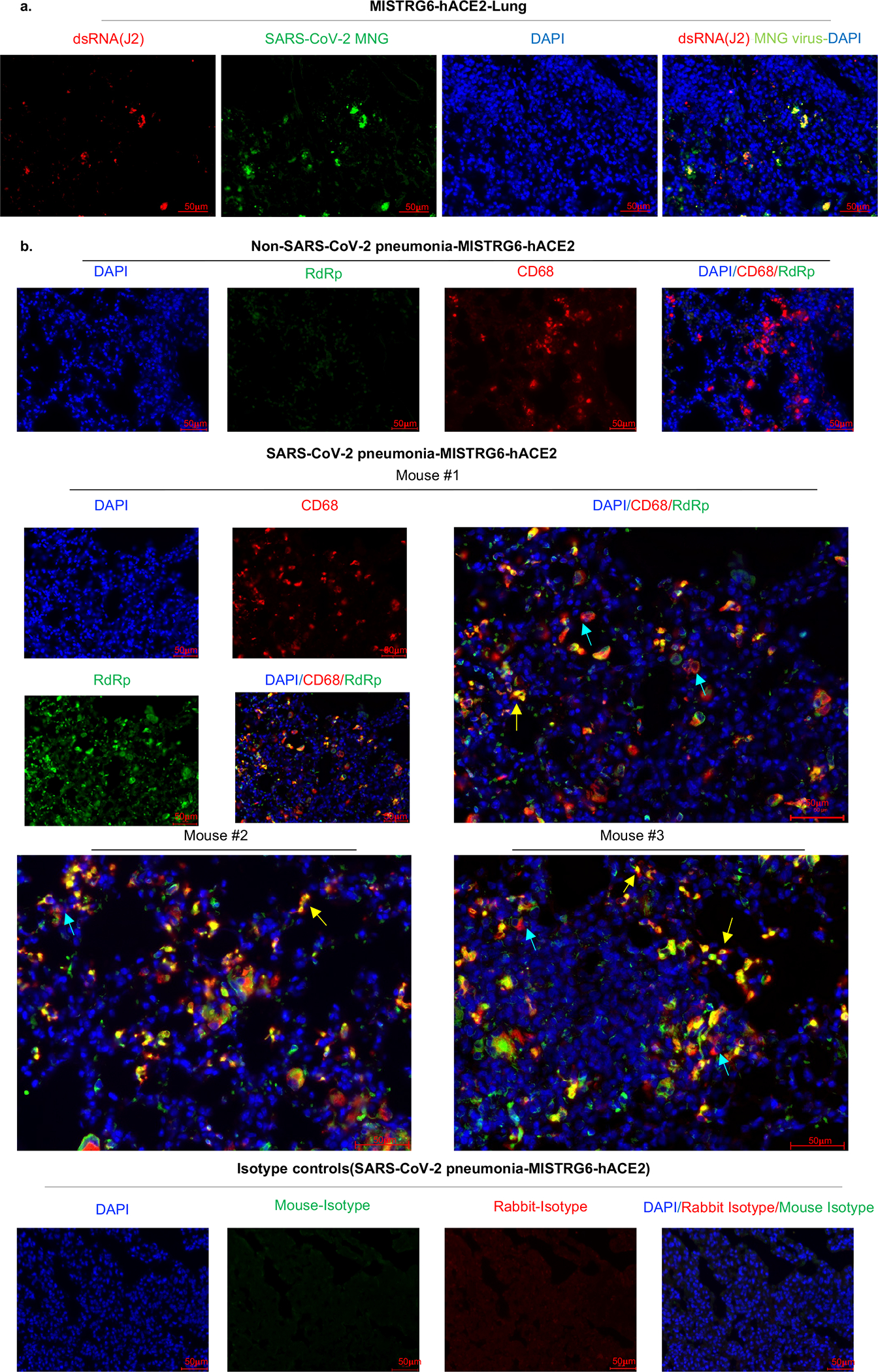
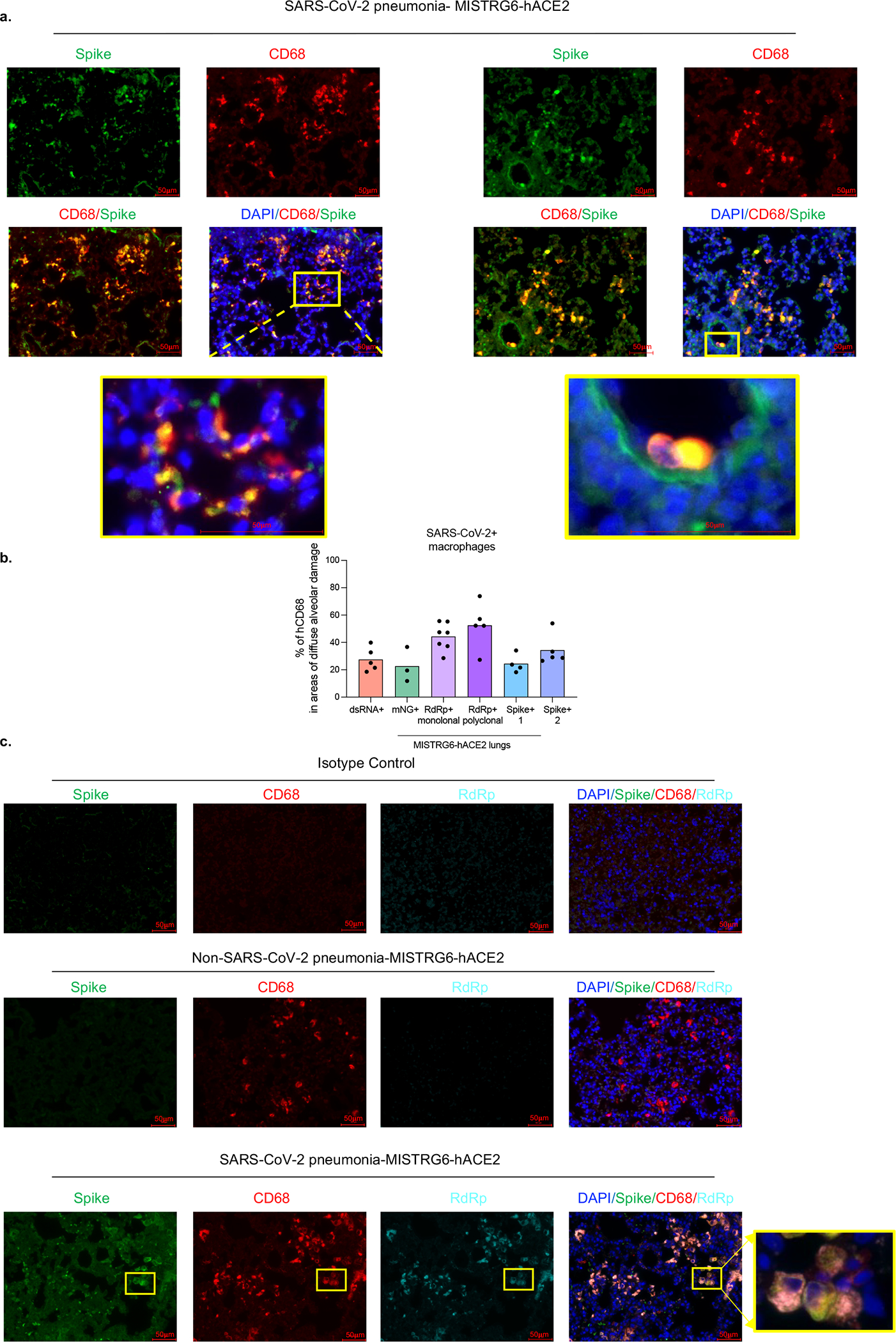
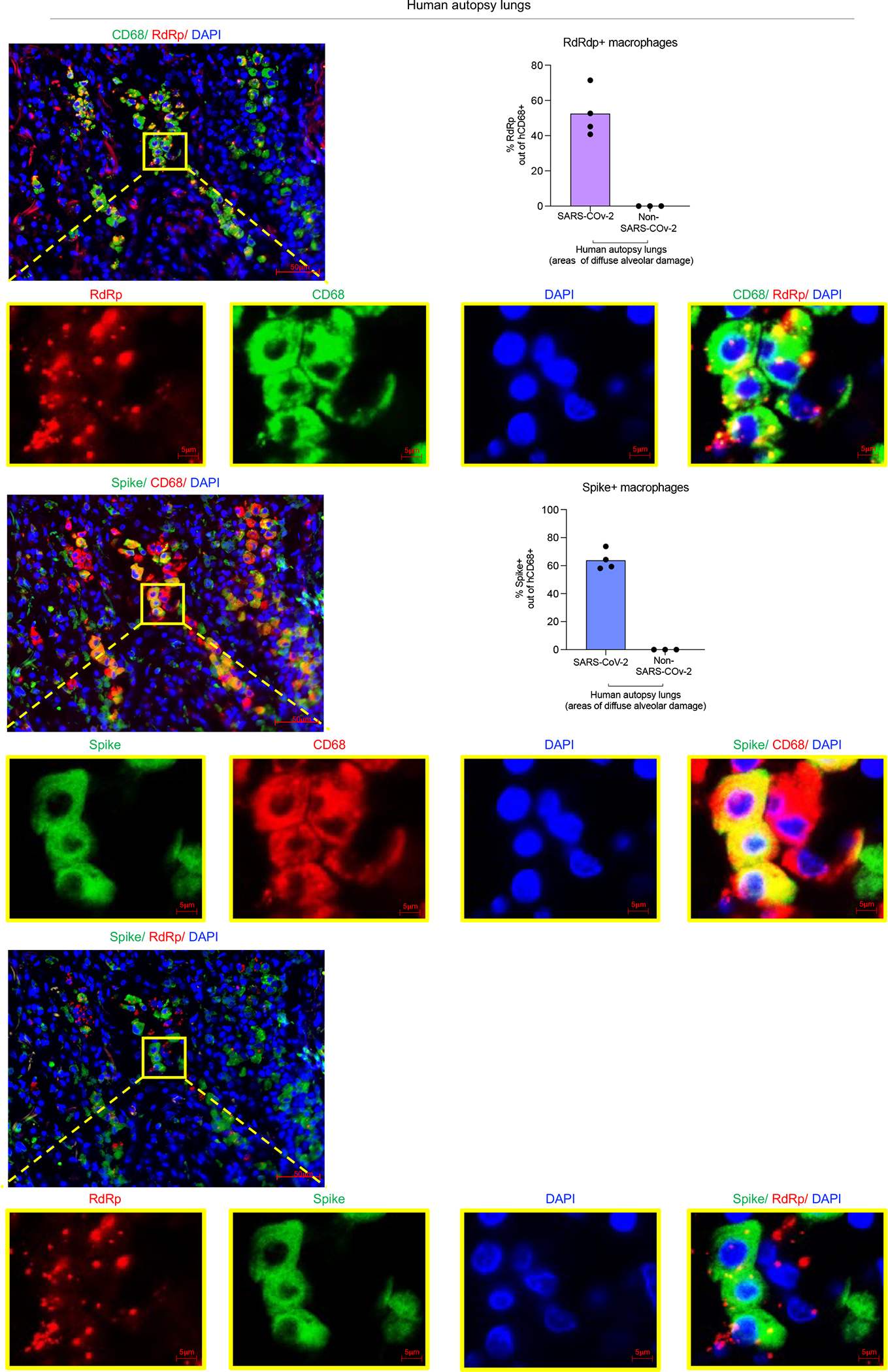
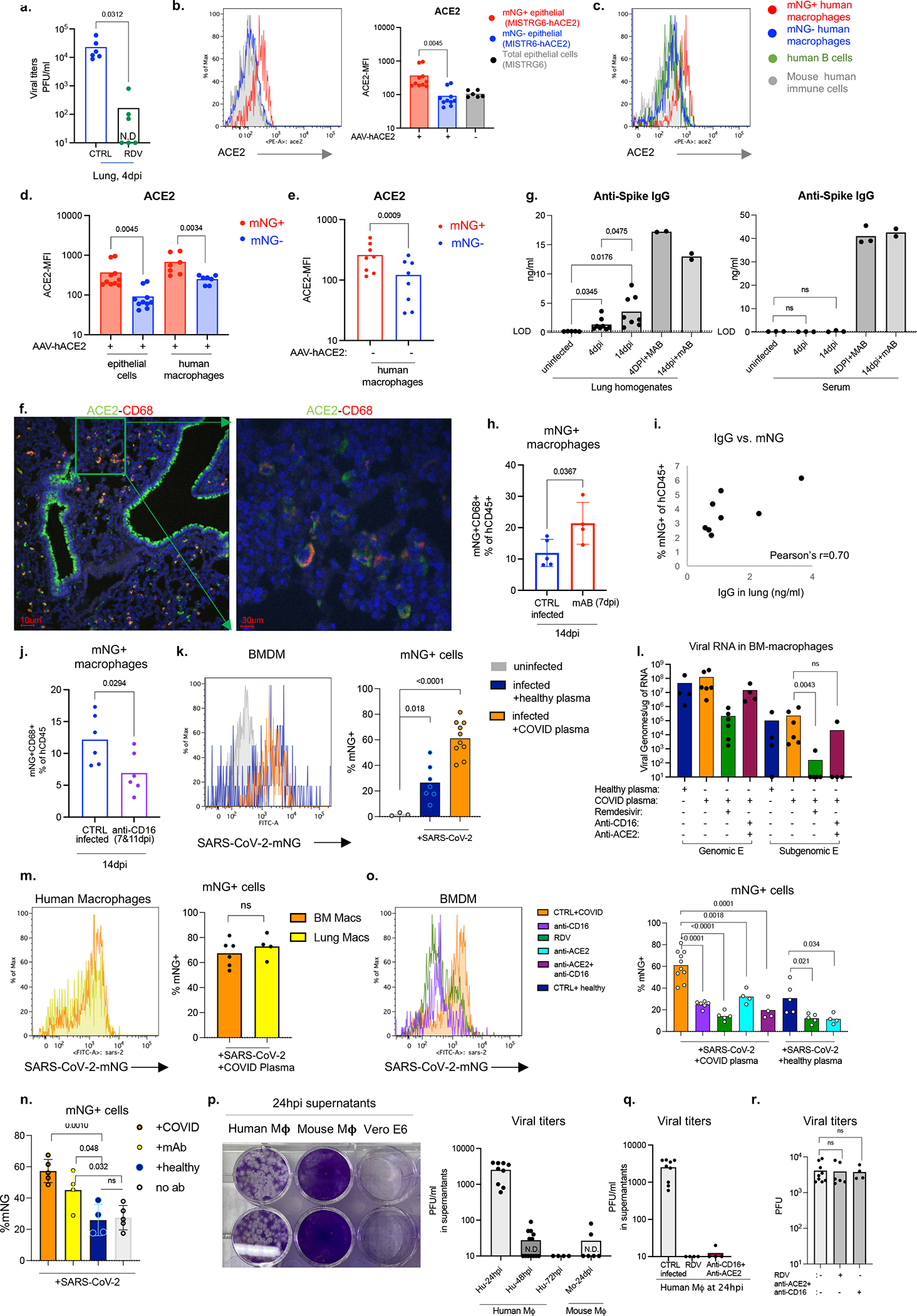
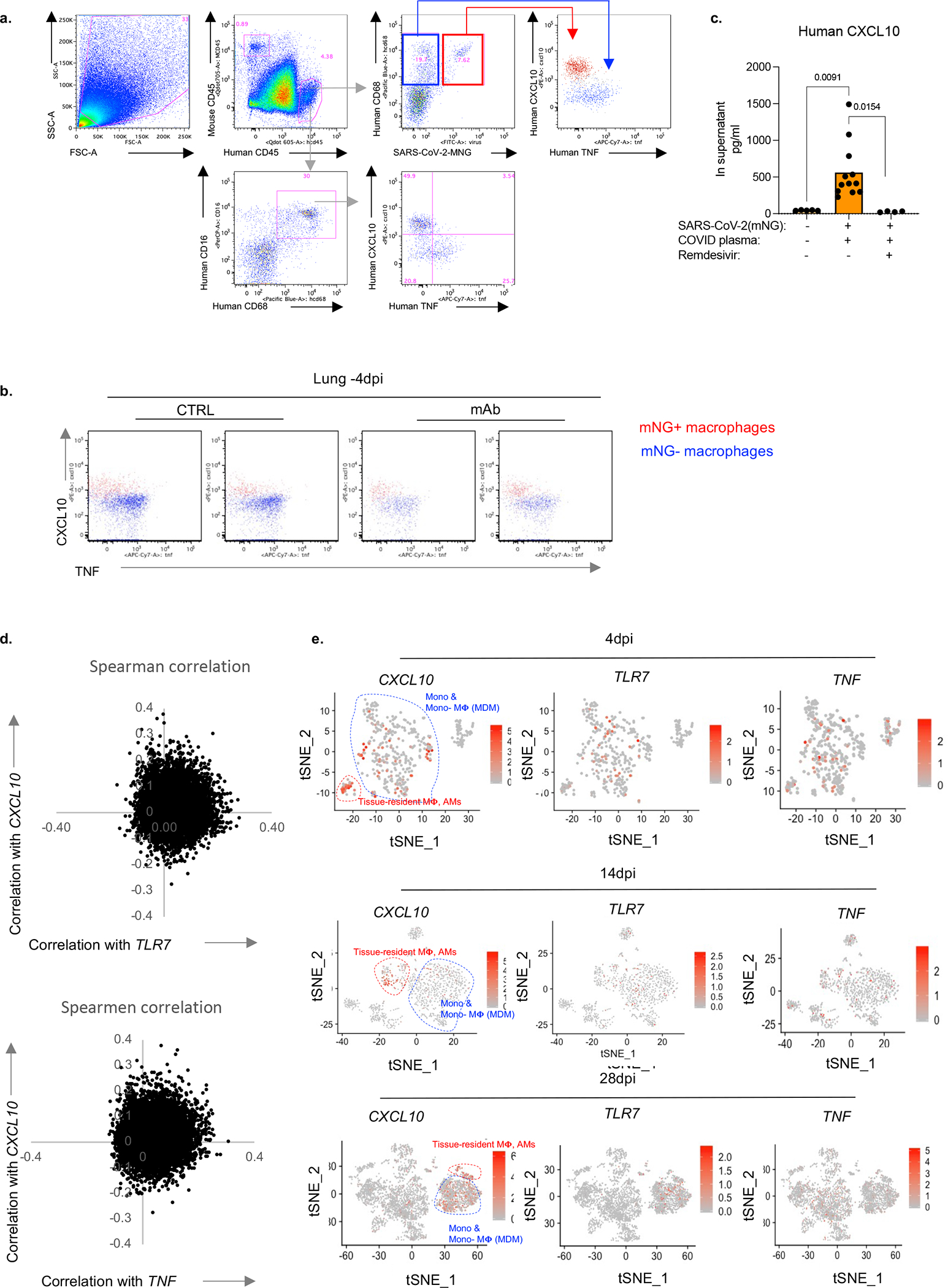
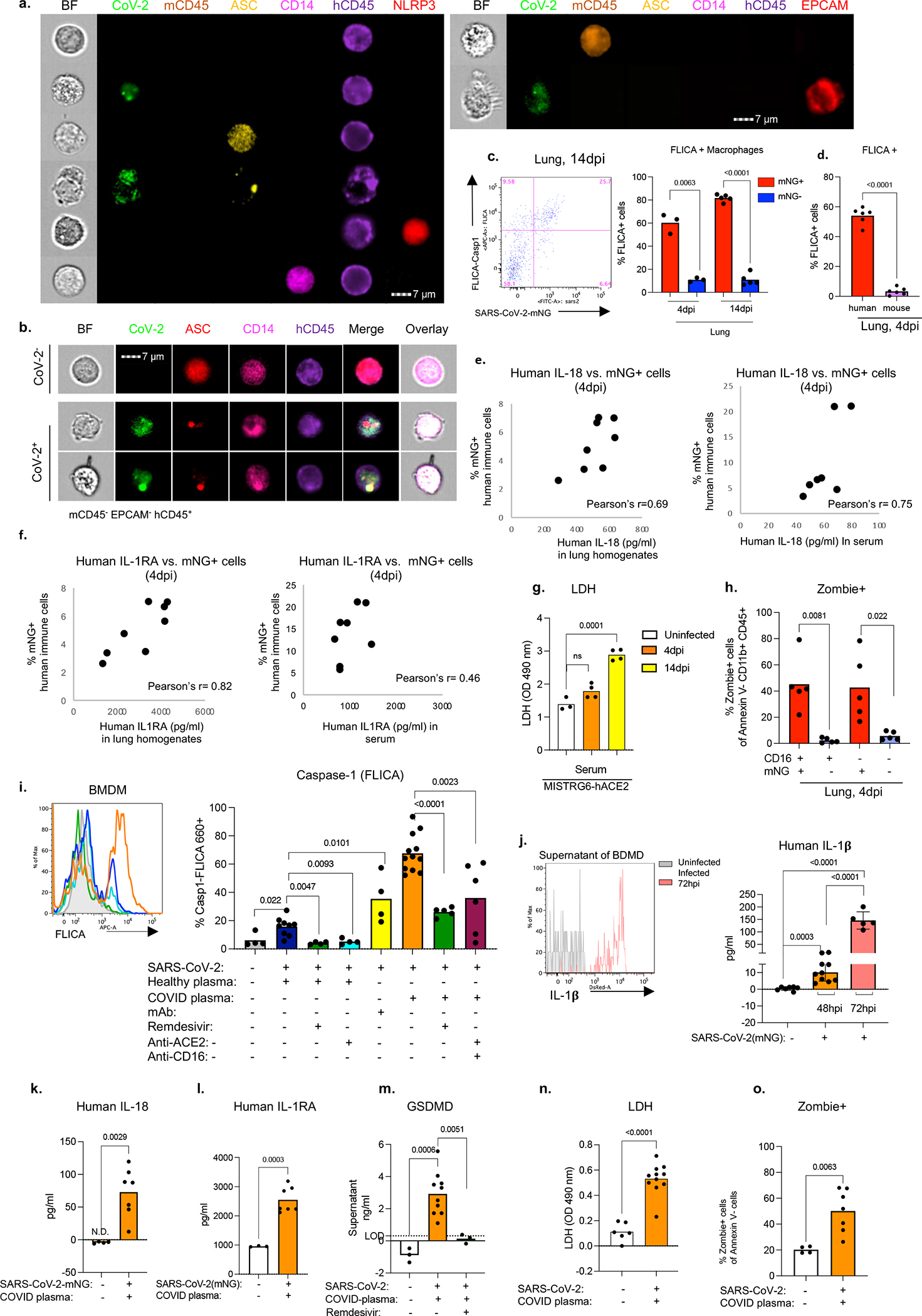
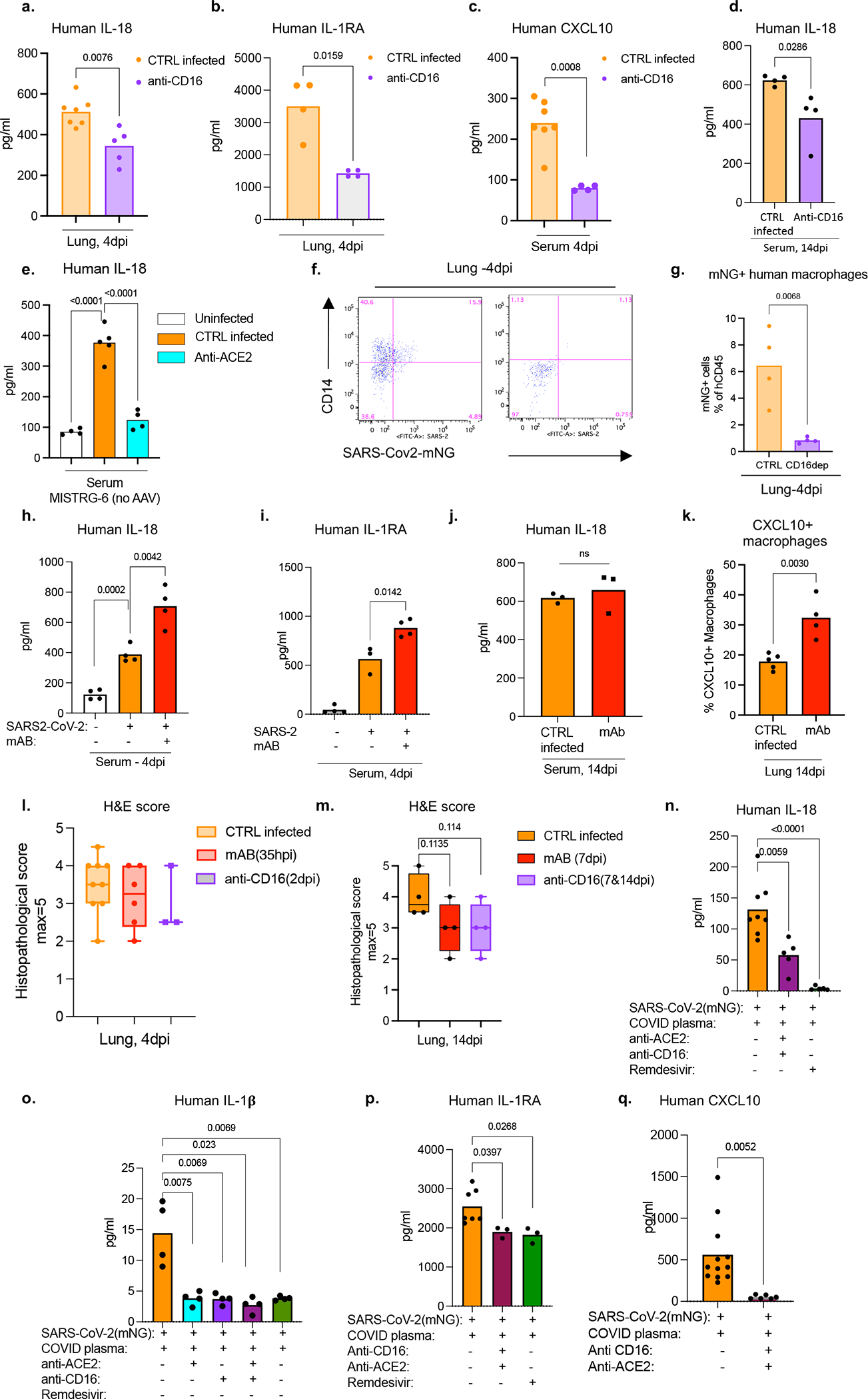
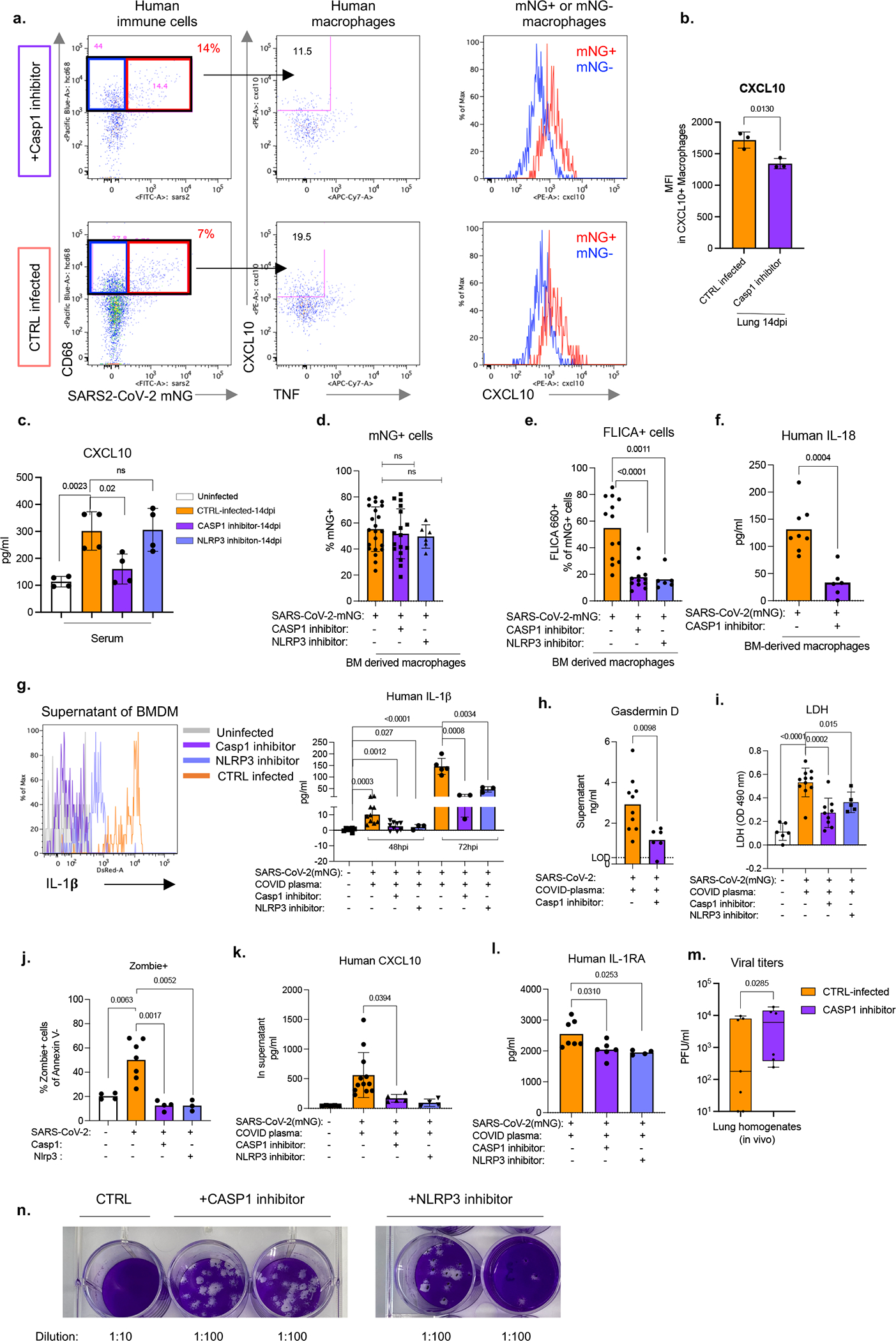
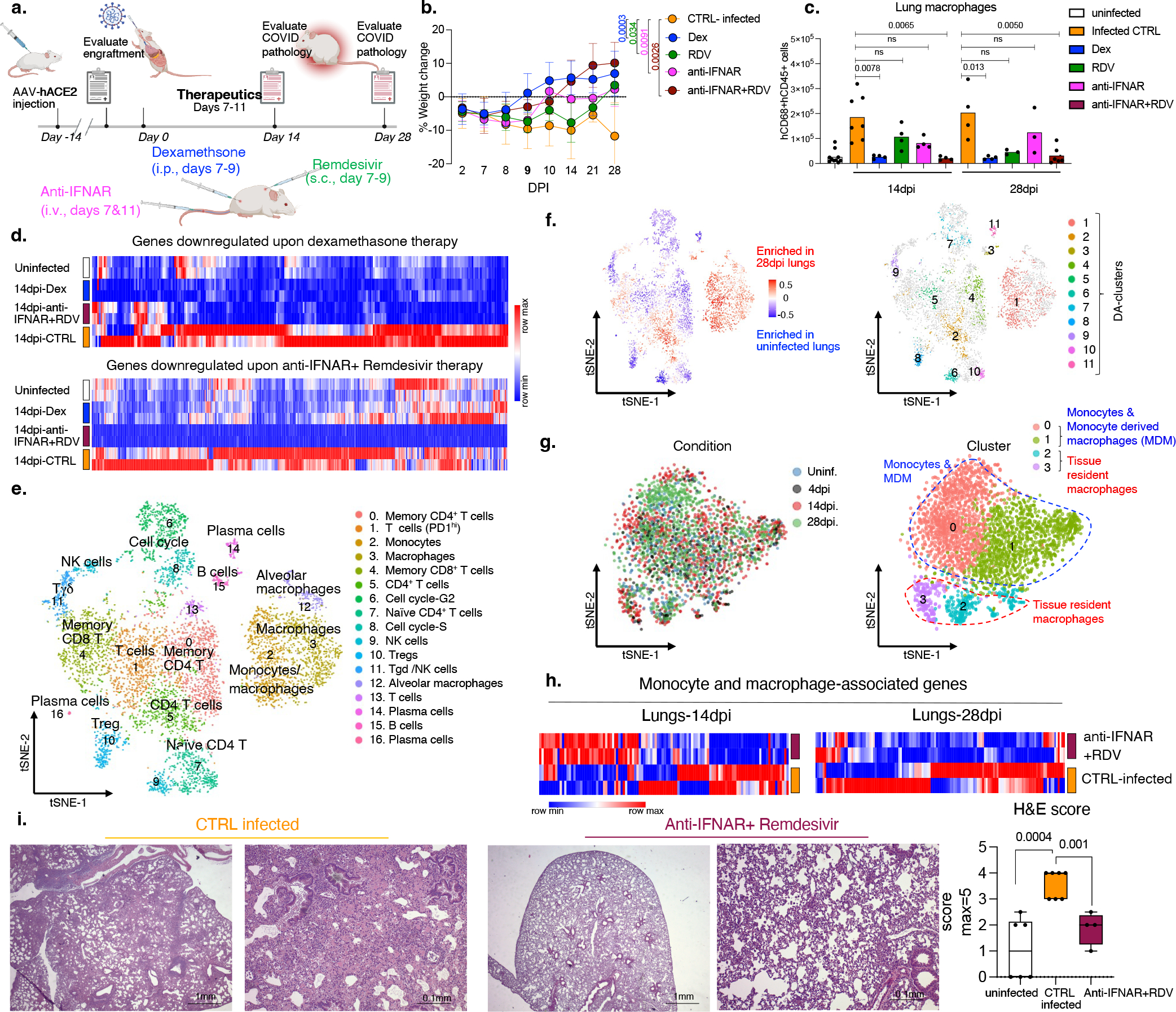
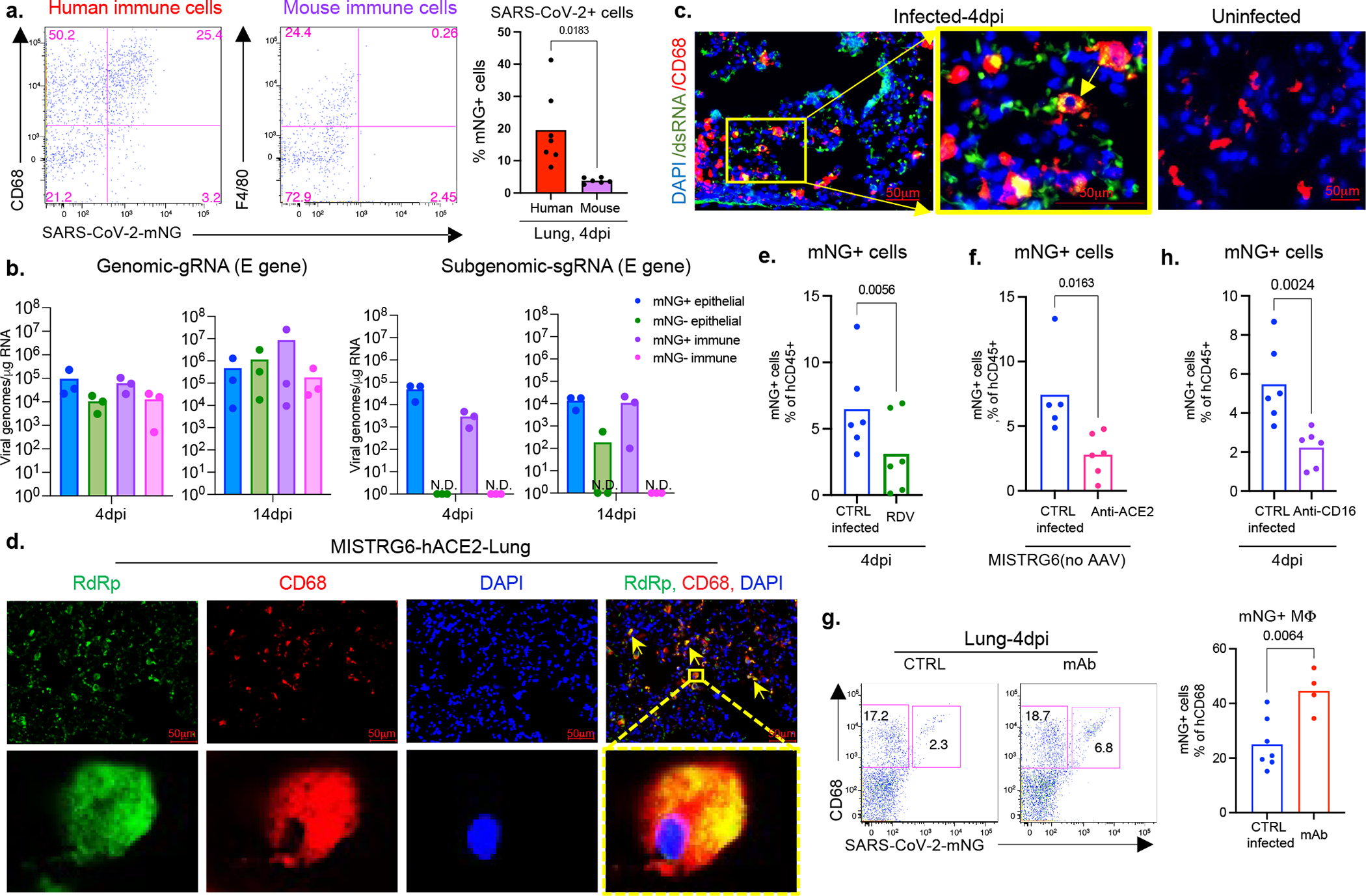
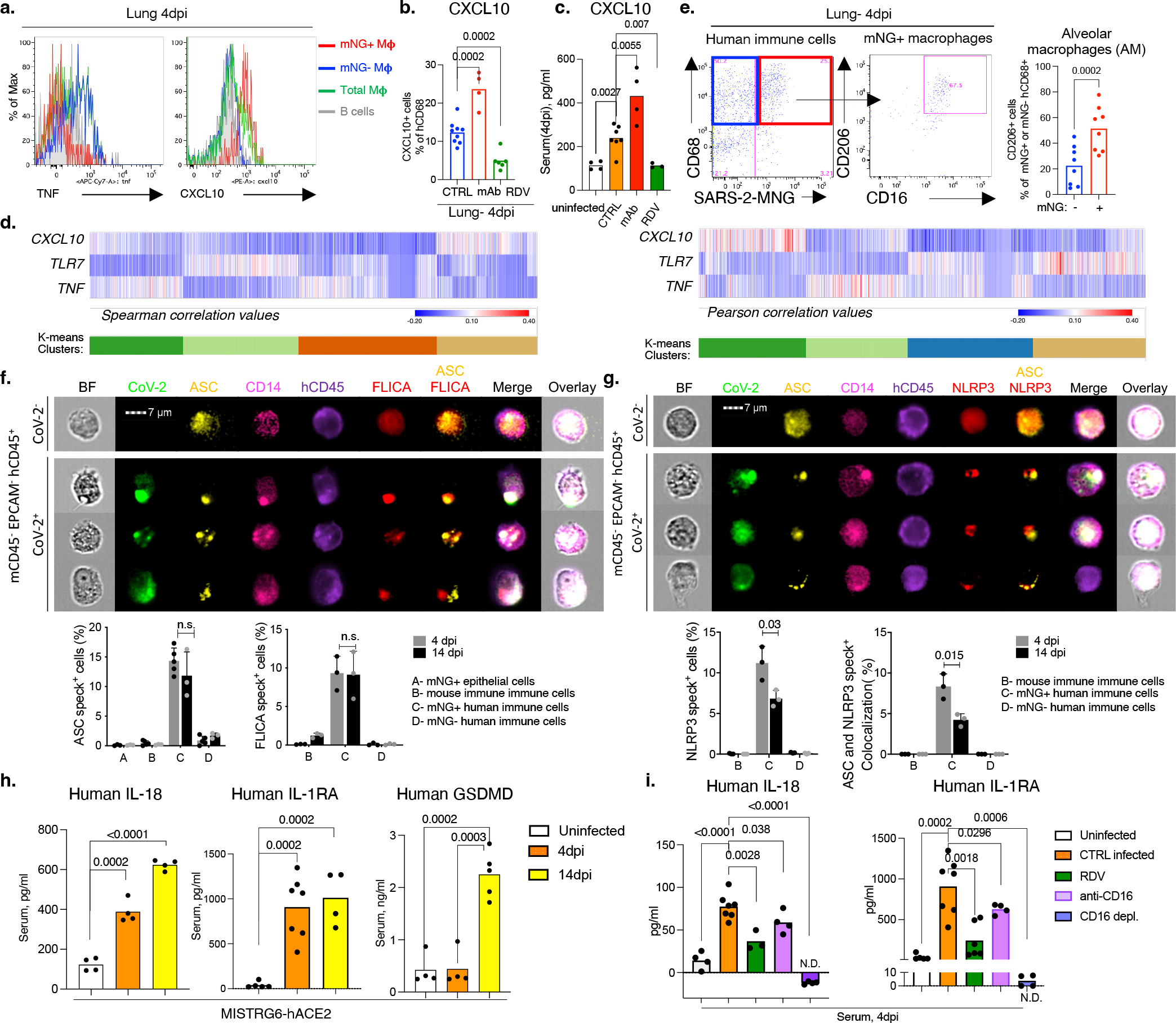
Severe COVID-19 is characterized by persistent lung inflammation, inflammatory cytokine production, viral RNA and a sustained interferon (IFN) response, all of which are recapitulated and required for pathology in the SARS-CoV-2-infected MISTRG6-hACE2 humanized mouse model of COVID-19, which has a human immune system. Blocking either viral replication with remdesivir or the downstream IFN-stimulated cascade with anti-IFNAR2 antibodies in vivo in the chronic stages of disease attenuates the overactive immune inflammatory response, especially inflammatory macrophages. Here we show that SARS-CoV-2 infection and replication in lung-resident human macrophages is a critical driver of disease. In response to infection mediated by CD16 and ACE2 receptors, human macrophages activate inflammasomes, release interleukin 1 (IL-1) and IL-18, and undergo pyroptosis, thereby contributing to the hyperinflammatory state of the lungs. Inflammasome activation and the accompanying inflammatory response are necessary for lung inflammation, as inhibition of the NLRP3 inflammasome pathway reverses chronic lung pathology. Notably, this blockade of inflammasome activation leads to the release of infectious virus by the infected macrophages. Thus, inflammasomes oppose host infection by SARS-CoV-2 through the production of inflammatory cytokines and suicide by pyroptosis to prevent a productive viral cycle.
严重的 COVID-19 的特征是持续的肺部炎症、炎症细胞因子的产生、病毒 RNA 和持续的干扰素 (IFN) 反应,所有这些都在 SARS-CoV-2 感染的 MISTRG6-hACE2 人类化小鼠 COVID-19 模型中得到重现和需要进行病理学研究,该模型具有人类免疫系统。在疾病的慢性阶段,用瑞德西韦阻断病毒复制或用抗 IFNAR2 抗体阻断下游 IFN 刺激级联反应,可减轻过度活跃的免疫炎症反应,尤其是炎症性巨噬细胞。在这里,我们表明,肺驻留的人类巨噬细胞中的 SARS-CoV-2 感染和复制是疾病的关键驱动因素。在 CD16 和 ACE2 受体介导的感染反应中,人类巨噬细胞激活炎性小体,释放白细胞介素 1 (IL-1) 和白细胞介素 18 (IL-18),并发生细胞焦亡,从而导致肺部的过度炎症状态。炎性小体的激活和伴随的炎症反应是肺部炎症所必需的,因为抑制 NLRP3 炎性小体途径可逆转慢性肺部病理学。值得注意的是,这种炎性小体激活的阻断会导致受感染的巨噬细胞释放传染性病毒。因此,炎性小体通过产生炎症细胞因子和细胞焦亡自杀来对抗 SARS-CoV-2 宿主感染,以防止病毒的有效复制周期。