Department of Neurology, Carver College of Medicine, University of Iowa, Iowa City, IA, 52242, USA.
InFlectis BioScience, 44300, Nantes, France.
Mol Neurobiol. 2022 Jul;59(7):4159-4178. doi: 10.1007/s12035-022-02838-y. Epub 2022 Apr 30.
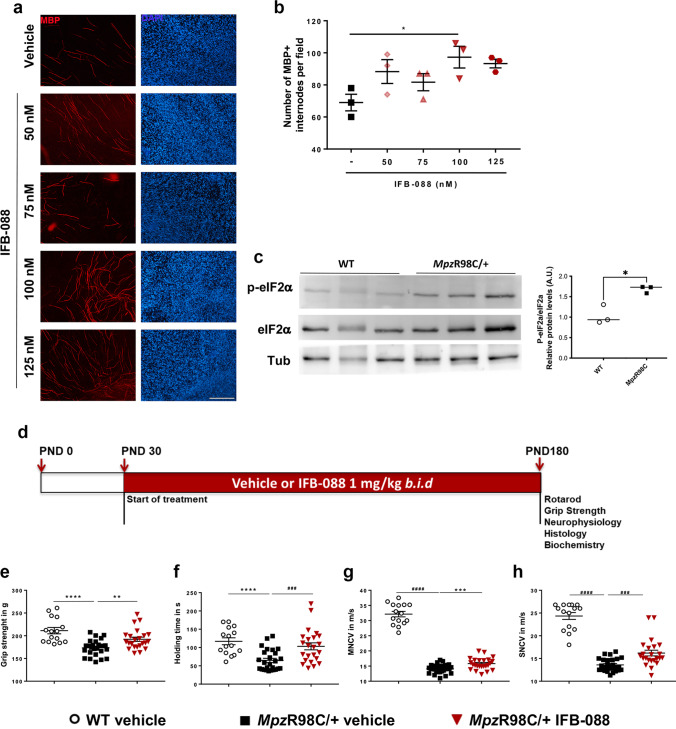
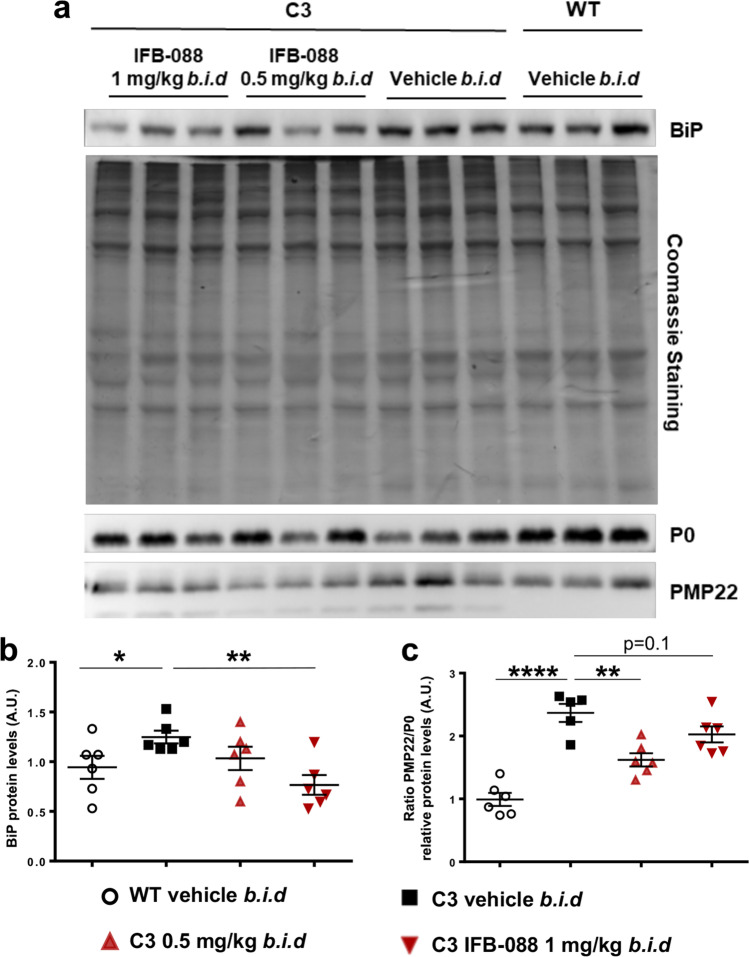
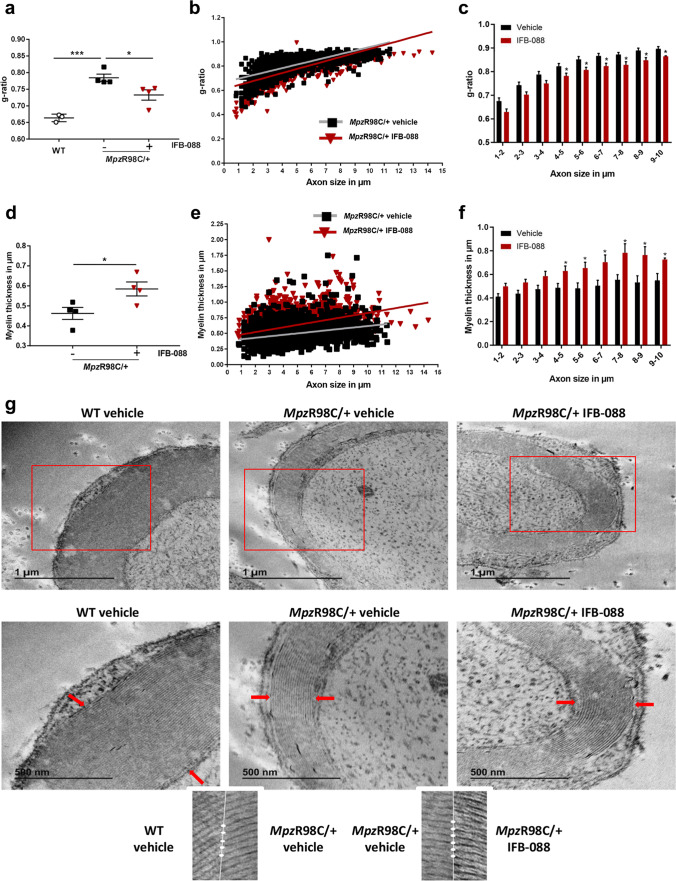
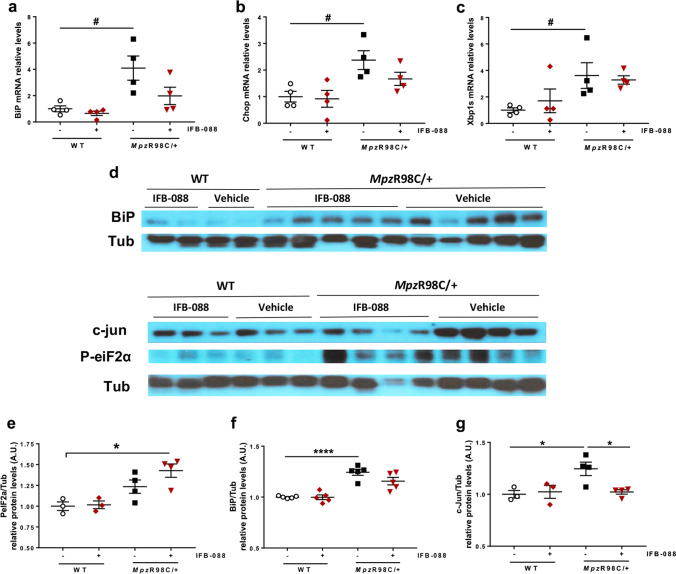
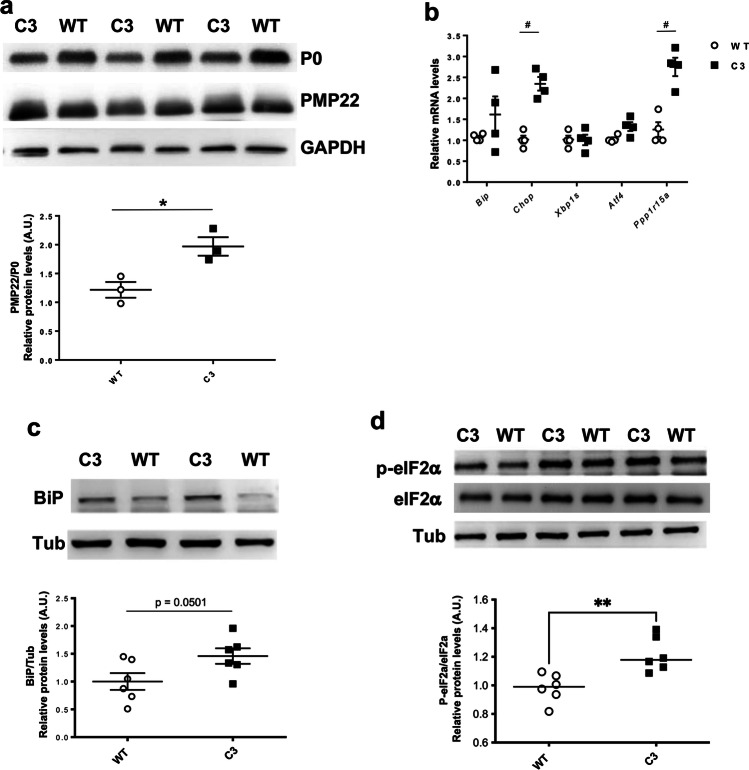
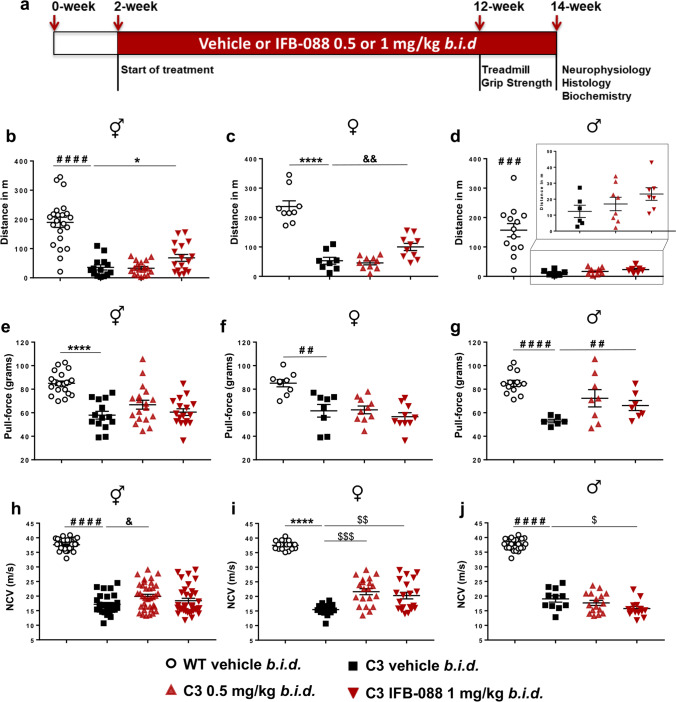
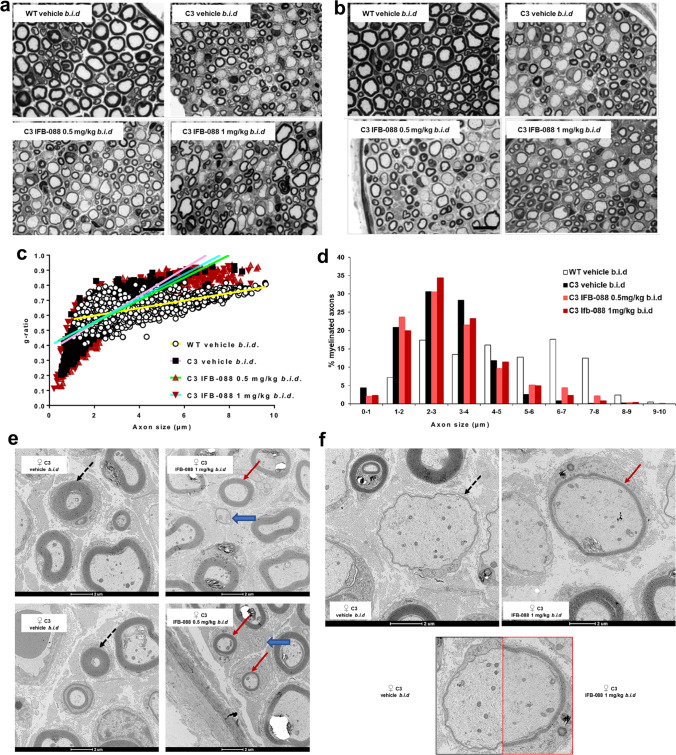
Charcot-Marie-Tooth disease type 1A (CMT1A), caused by duplication of the peripheral myelin protein 22 (PMP22) gene, and CMT1B, caused by mutations in myelin protein zero (MPZ) gene, are the two most common forms of demyelinating CMT (CMT1), and no treatments are available for either. Prior studies of the MpzSer63del mouse model of CMT1B have demonstrated that protein misfolding, endoplasmic reticulum (ER) retention and activation of the unfolded protein response (UPR) contributed to the neuropathy. Heterozygous patients with an arginine to cysteine mutation in MPZ (MPZR98C) develop a severe infantile form of CMT1B which is modelled by MpzR98C/ + mice that also show ER stress and an activated UPR. C3-PMP22 mice are considered to effectively model CMT1A. Altered proteostasis, ER stress and activation of the UPR have been demonstrated in mice carrying Pmp22 mutations. To determine whether enabling the ER stress/UPR and readjusting protein homeostasis would effectively treat these models of CMT1B and CMT1A, we administered Sephin1/IFB-088/icerguestat, a UPR modulator which showed efficacy in the MpzS63del model of CMT1B, to heterozygous MpzR98C and C3-PMP22 mice. Mice were analysed by behavioural, neurophysiological, morphological and biochemical measures. Both MpzR98C/ + and C3-PMP22 mice improved in motor function and neurophysiology. Myelination, as demonstrated by g-ratios and myelin thickness, improved in CMT1B and CMT1A mice and markers of UPR activation returned towards wild-type values. Taken together, our results demonstrate the capability of IFB-088 to treat a second mouse model of CMT1B and a mouse model of CMT1A, the most common form of CMT. Given the recent benefits of IFB-088 treatment in amyotrophic lateral sclerosis and multiple sclerosis animal models, these data demonstrate its potential in managing UPR and ER stress for multiple mutations in CMT1 as well as in other neurodegenerative diseases. (Left panel) the accumulation of overexpressed PMP22 or misfolded mutant P0 in the Schwann cell endoplasmic reticulum (ER) leads to overwhelming of the degradative capacity, activation of ER-stress mechanisms, and myelination impairment. (Right panel) by prolonging eIF2α phosphorylation, IFB-088 reduces the amount of newly synthesized proteins entering the ER, allowing the protein quality control systems to better cope with the unfolded/misfolded protein and allowing myelination to progress.
腓骨肌萎缩症 1A 型(CMT1A)由外周髓鞘蛋白 22(PMP22)基因重复引起,CMT1B 由髓鞘蛋白零(MPZ)基因突变引起,是脱髓鞘性 CMT(CMT1)的两种最常见形式,目前均尚无治疗方法。先前对 CMT1B 的 MpzSer63del 小鼠模型的研究表明,蛋白质错误折叠、内质网(ER)保留和未折叠蛋白反应(UPR)的激活导致了神经病变。MPZ 中精氨酸到半胱氨酸突变的杂合子患者会发展为严重的婴儿期 CMT1B,MpzR98C/ + 小鼠可对此进行建模,该模型也表现出 ER 应激和激活的 UPR。C3-PMP22 小鼠被认为可有效模拟 CMT1A。在携带 Pmp22 突变的小鼠中,已经证明了蛋白质平衡、ER 应激和 UPR 的激活。为了确定是否通过激活 ER 应激/UPR 和重新调整蛋白质稳态可以有效治疗 CMT1B 和 CMT1A 这些模型,我们向杂合子 MpzR98C 和 C3-PMP22 小鼠给予 Sephin1/IFB-088/icerguestat,一种在 CMT1B 的 MpzS63del 模型中表现出疗效的 UPR 调节剂。通过行为、神经生理学、形态和生化测量来分析小鼠。MpzR98C/ + 和 C3-PMP22 小鼠的运动功能和神经生理学均得到改善。CMT1B 和 CMT1A 小鼠的髓鞘化(由 g-比和髓鞘厚度表明)得到改善,UPR 激活的标志物恢复到野生型水平。总的来说,我们的结果表明 IFB-088 能够治疗 CMT1B 的第二种小鼠模型和 CMT1A 的小鼠模型,这是 CMT 最常见的形式。鉴于 IFB-088 最近在肌萎缩侧索硬化症和多发性硬化症动物模型中的益处,这些数据表明其在管理 CMT1 中多种突变以及其他神经退行性疾病中的 UPR 和 ER 应激方面具有潜力。(左图)过表达的 PMP22 或突变的 P0 错误折叠在施万细胞内质网(ER)中的积累导致降解能力过载,激活 ER 应激机制,并导致髓鞘形成受损。(右图)通过延长 eIF2α 磷酸化,IFB-088 减少进入 ER 的新合成蛋白质的量,使蛋白质质量控制系统更好地应对未折叠/错误折叠的蛋白质,并允许髓鞘形成继续进行。