Olivia Newton-John Cancer and Research Institute, Melbourne, VIC, Australia.
School of Cancer Medicine, La Trobe University, Melbourne, VIC, Australia.
BMC Cancer. 2022 May 2;22(1):478. doi: 10.1186/s12885-022-09478-4.
Mutations and fusions in Fibroblast Growth Factor Receptor 3 (FGFR3) occur in 10-20% of metastatic urothelial carcinomas and confer sensitivity to FGFR inhibitors. However, responses to these agents are often short-lived due to the development of acquired resistance. The objective of this study was to identify mechanisms of resistance to FGFR inhibitors in two previously uncharacterised bladder cancer cell lines harbouring FGFR3 fusions and assess rational combination therapies to enhance sensitivity to these agents.
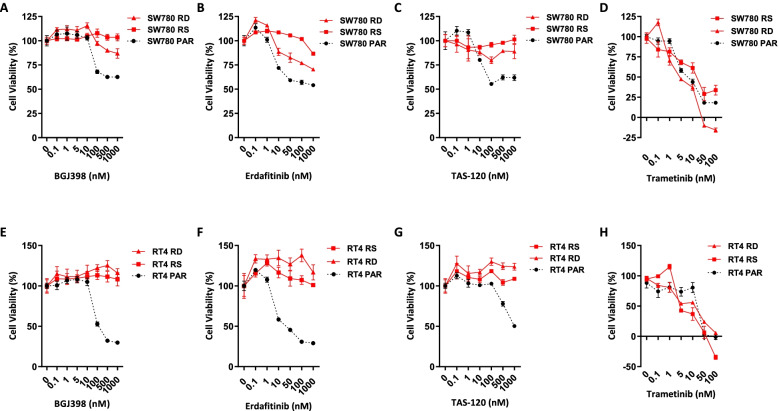
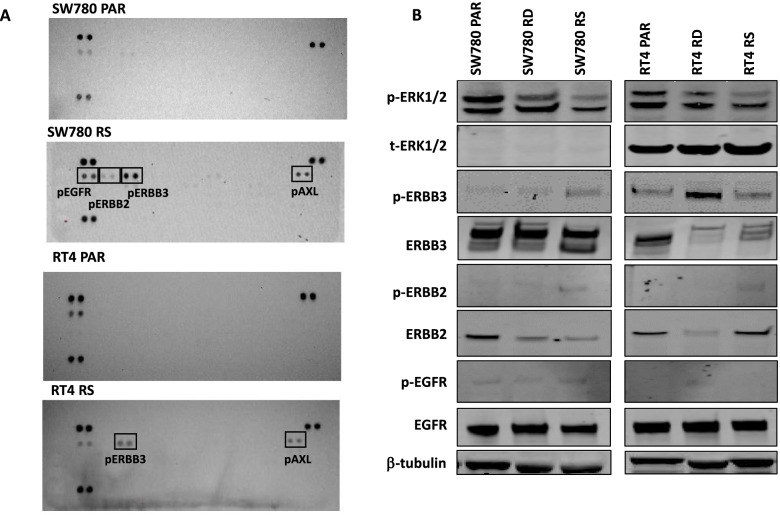
Acquired resistance to FGFR inhibitors was generated in two FGFR3 fusion harbouring cell lines, SW780 (FGFR3-BAIAP2L1 fusion) and RT4 (FGFR3-TACC3 fusion), by long-term exposure to the FGFR inhibitor BGJ398. Changes in levels of receptor tyrosine kinases were assessed by phospho-RTK arrays and immunoblotting. Changes in cell viability and proliferation were assessed by the Cell-Titre Glo assay and by propidium iodide staining and FACS analysis.
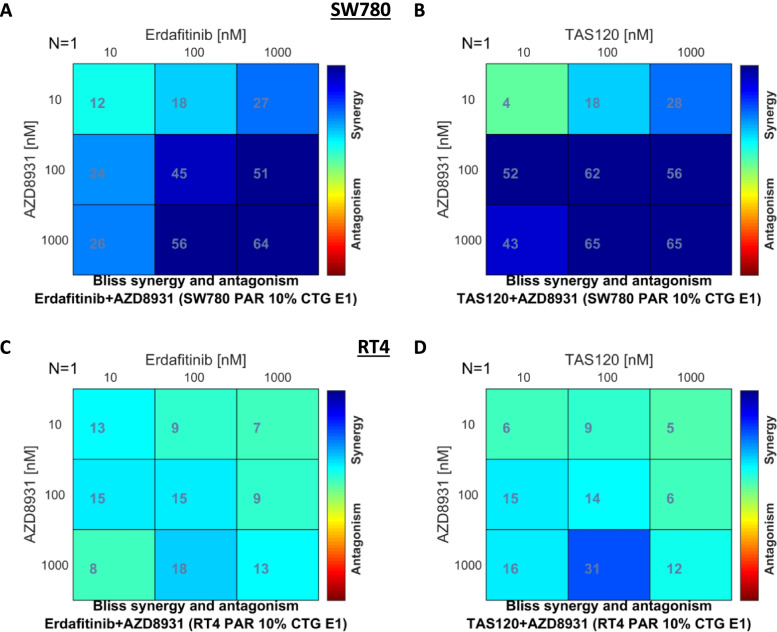
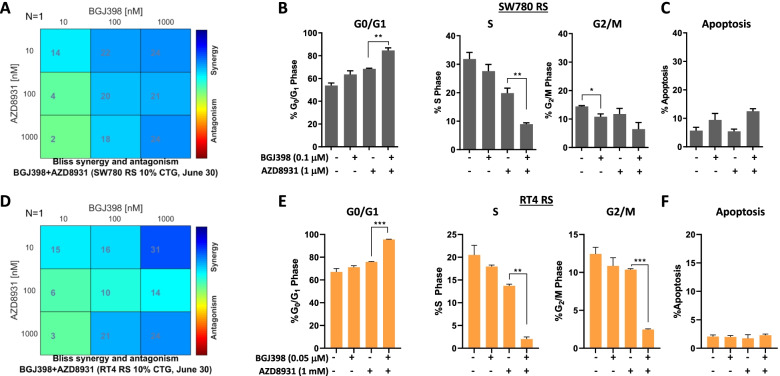
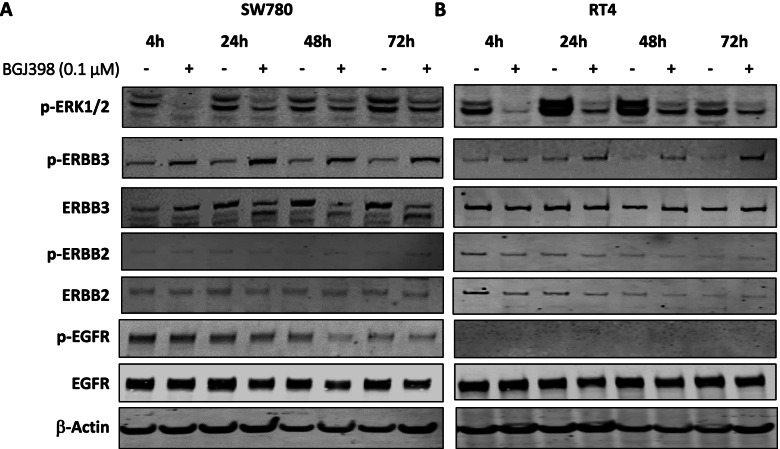
Long term treatment of FGFR3-fusion harbouring SW780 and RT4 bladder cancer cell lines with the FGFR inhibitor BGJ398 resulted in the establishment of resistant clones. These clones were cross-resistant to the clinically approved FGFR inhibitor erdafitinib and the covalently binding irreversible FGFR inhibitor TAS-120, but remained sensitive to the MEK inhibitor trametinib, indicating resistance is mediated by alternate activation of MAPK signalling. The FGFR inhibitor-resistant SW780 and RT4 lines displayed increased expression of pERBB3, and strikingly, combination treatment with an FGFR inhibitor and the ATP-competitive pan-ERBB inhibitor AZD8931 overcame this resistance. Notably, rapid induction of pERBB3 and reactivation of pERK also occurred in parental FGFR3 fusion-driven lines within 24 h of FGFR inhibitor treatment, and combination treatment with an FGFR inhibitor and AZD8931 delayed the reactivation of pERBB3 and pERK and synergistically inhibited cell proliferation.
We demonstrate that increased expression of pERBB3 is a key mechanism of adaptive resistance to FGFR inhibitors in FGFR3-fusion driven bladder cancers, and that this also occurs rapidly following FGFR inhibitor treatment. Our findings demonstrate that resistance can be overcome by combination treatment with a pan-ERBB inhibitor and suggest that upfront combination treatment with FGFR and pan-ERBB inhibitors warrants further investigation for FGFR3-fusion harbouring bladder cancers.
成纤维细胞生长因子受体 3(FGFR3)的突变和融合发生在 10-20%的转移性尿路上皮癌中,并对 FGFR 抑制剂敏感。然而,由于获得性耐药的发展,这些药物的反应往往是短暂的。本研究的目的是在两个以前未被描述的膀胱癌细胞系中鉴定 FGFR 抑制剂耐药的机制,这些细胞系中存在 FGFR3 融合,并评估合理的联合治疗方法来增强对这些药物的敏感性。
通过长期暴露于 FGFR 抑制剂 BGJ398,在两个 FGFR3 融合阳性细胞系 SW780(FGFR3-BAIAP2L1 融合)和 RT4(FGFR3-TACC3 融合)中产生对 FGFR 抑制剂的获得性耐药。通过磷酸化 RTK 阵列和免疫印迹评估受体酪氨酸激酶水平的变化。通过细胞 - 荧光素酶测定法和碘化丙啶染色和 FACS 分析评估细胞活力和增殖的变化。
长期用 FGFR 抑制剂 BGJ398 处理 FGFR3 融合阳性的 SW780 和 RT4 膀胱癌细胞系导致建立了耐药克隆。这些克隆对临床批准的 FGFR 抑制剂 erdafitinib 和共价结合的不可逆 FGFR 抑制剂 TAS-120 具有交叉耐药性,但对 MEK 抑制剂 trametinib 仍敏感,表明耐药性是由 MAPK 信号通路的替代激活介导的。FGFR 抑制剂耐药的 SW780 和 RT4 株显示出 pERBB3 的表达增加,引人注目的是,FGFR 抑制剂与 ATP 竞争性泛 ERBB 抑制剂 AZD8931 的联合治疗克服了这种耐药性。值得注意的是,在 FGFR 抑制剂治疗后 24 小时内,FGFR3 融合驱动的亲本系中也迅速诱导 pERBB3 的表达,并重新激活 pERK,并且 FGFR 抑制剂与 AZD8931 的联合治疗延迟了 pERBB3 和 pERK 的再激活,并协同抑制细胞增殖。
我们证明,pERBB3 的表达增加是 FGFR3 融合驱动的膀胱癌对 FGFR 抑制剂获得性耐药的关键机制,并且在 FGFR 抑制剂治疗后也迅速发生。我们的研究结果表明,通过联合使用泛 ERBB 抑制剂可以克服耐药性,并表明在 FGFR3 融合阳性膀胱癌中,联合使用 FGFR 和泛 ERBB 抑制剂进行前期治疗值得进一步研究。