Babu Sathya, Nagarajan Santhosh Kumar, Sathish Sruthy, Negi Vir Singh, Sohn Honglae, Madhavan Thirumurthy
Computational Biology Lab, Department of Genetic Engineering, School of Bioengineering, SRM Institute of Science and Technology, SRM Nagar, Kattankulathur, India.
Department of Clinical Immunology, Jawaharlal Institute of Post-Graduate Medical Education and Research, Pondicherry, India.
Front Pharmacol. 2022 Apr 21;13:837369. doi: 10.3389/fphar.2022.837369. eCollection 2022.
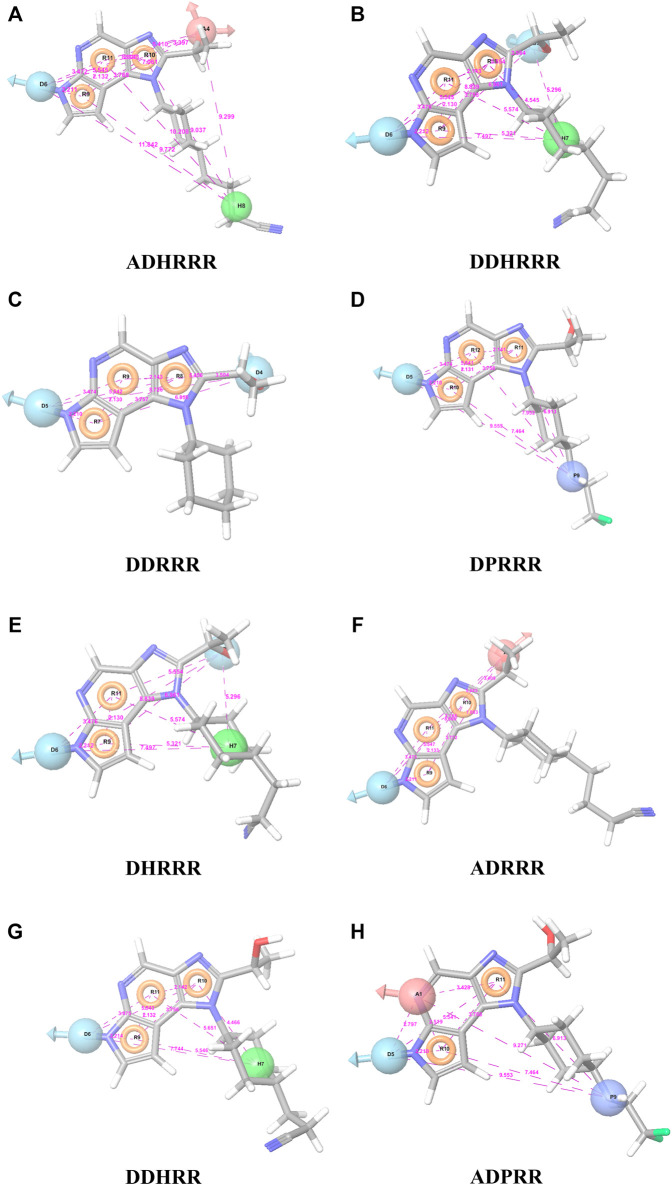
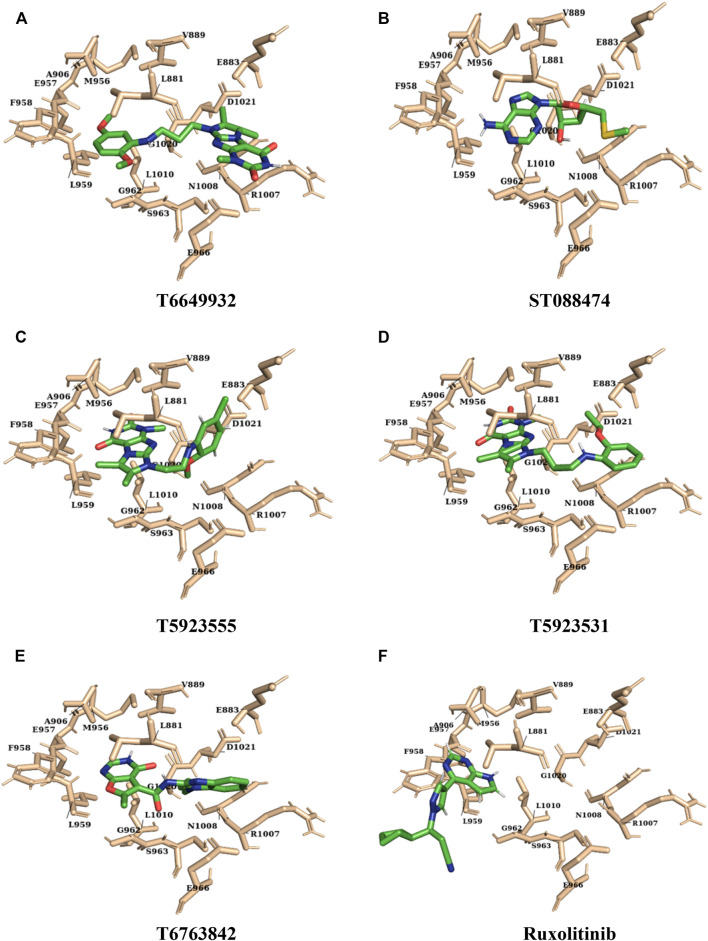
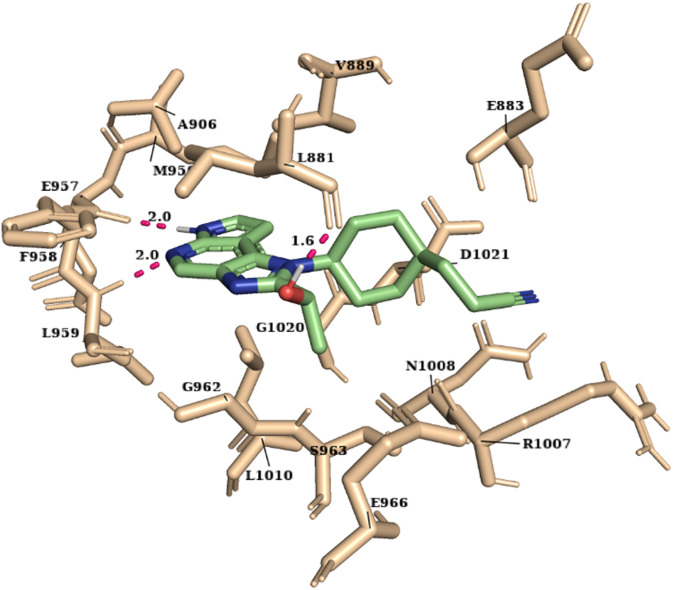
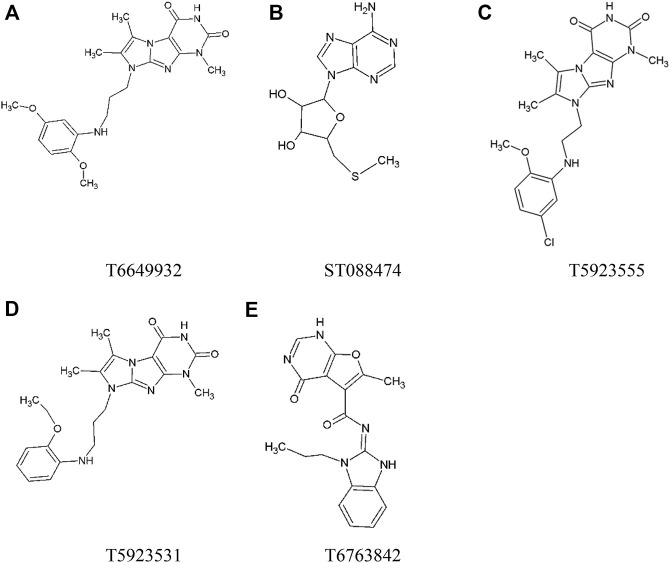
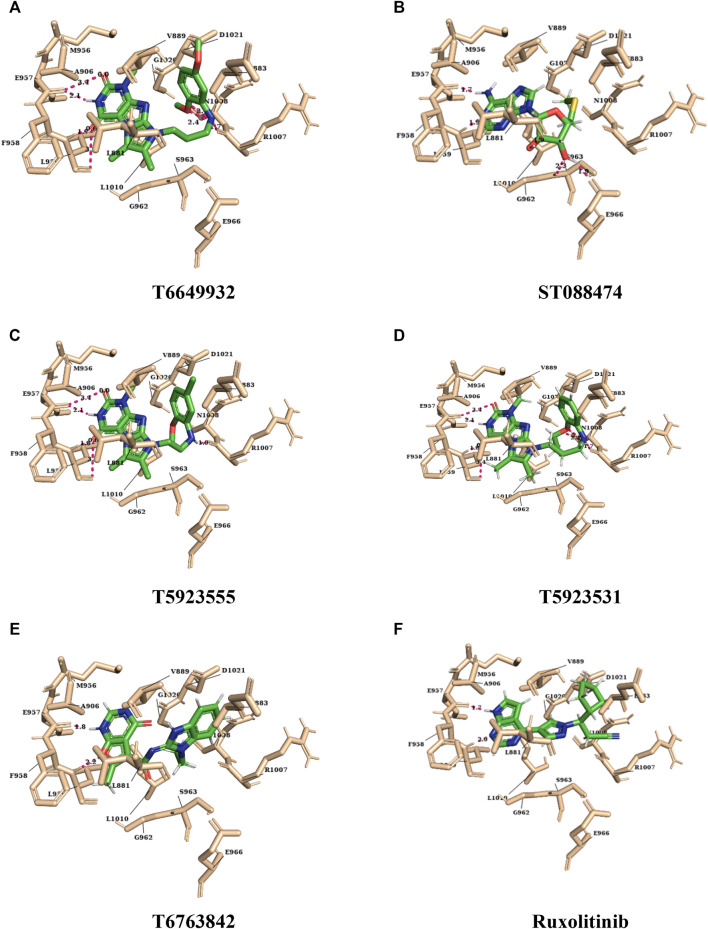
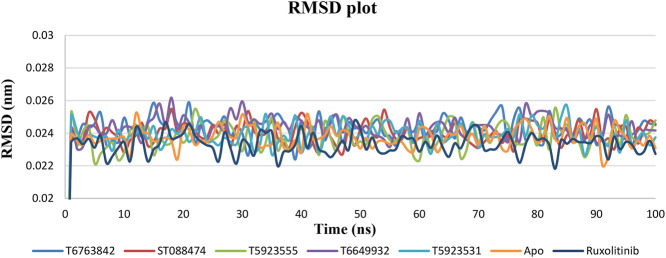
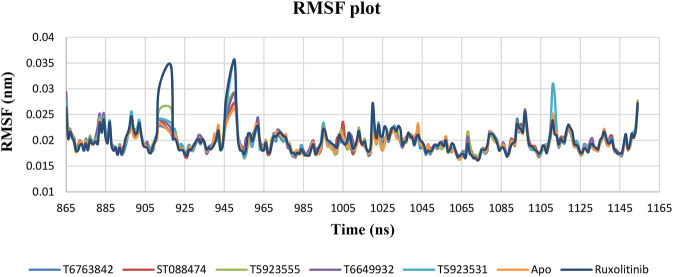
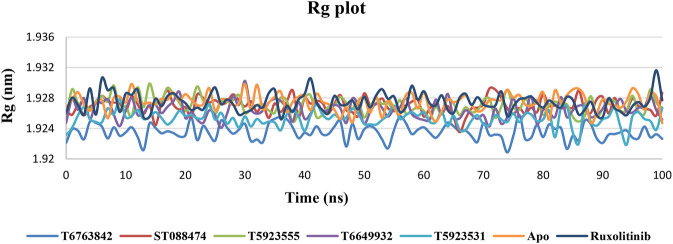
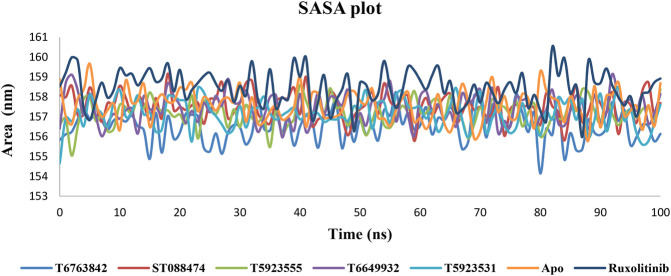
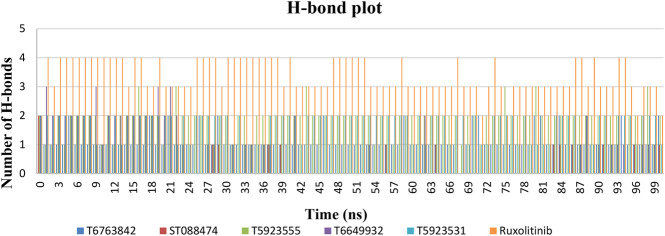
JAK1 plays a significant role in the intracellular signaling by interacting with cytokine receptors in different types of cells and is linked to the pathogenesis of various cancers and in the pathology of the immune system. In this study, ligand-based pharmacophore modeling combined with virtual screening and molecular docking methods was incorporated to identify the potent and selective lead compounds for JAK1. Initially, the ligand-based pharmacophore models were generated using a set of 52 JAK1 inhibitors named C-2 methyl/hydroxyethyl imidazopyrrolopyridines derivatives. Twenty-seven pharmacophore models with five and six pharmacophore features were generated and validated using potency and selectivity validation methods. During potency validation, the Guner-Henry score was calculated to check the accuracy of the generated models, whereas in selectivity validation, the pharmacophore models that are capable of identifying selective JAK1 inhibitors were evaluated. Based on the validation results, the best pharmacophore models ADHRRR, DDHRRR, DDRRR, DPRRR, DHRRR, ADRRR, DDHRR, and ADPRR were selected and taken for virtual screening against the Maybridge, Asinex, Chemdiv, Enamine, Lifechemicals, and Zinc database to identify the new molecules with novel scaffold that can bind to JAK1. A total of 4,265 hits were identified from screening and checked for acceptable drug-like properties. A total of 2,856 hits were selected after ADME predictions and taken for Glide molecular docking to assess the accurate binding modes of the lead candidates. Ninety molecules were shortlisted based on binding energy and H-bond interactions with the important residues of JAK1. The docking results were authenticated by calculating binding free energy for protein-ligand complexes using the MM-GBSA calculation and induced fit docking methods. Subsequently, the cross-docking approach was carried out to recognize the selective JAK1 lead compounds. Finally, top five lead compounds that were potent and selective against JAK1 were selected and validated using molecular dynamics simulation. Besides, the density functional theory study was also carried out for the selected leads. Through various computational studies, we observed good potency and selectivity of these lead compounds when compared with the drug ruxolitinib. Compounds such as T5923555 and T5923531 were found to be the best and can be further validated using and methods.
JAK1通过与不同类型细胞中的细胞因子受体相互作用,在细胞内信号传导中发挥重要作用,并与多种癌症的发病机制以及免疫系统的病理学相关。在本研究中,结合基于配体的药效团建模、虚拟筛选和分子对接方法,以鉴定JAK1的强效和选择性先导化合物。最初,使用一组名为C-2甲基/羟乙基咪唑并吡咯并吡啶衍生物的52种JAK1抑制剂生成基于配体的药效团模型。生成了27个具有五个和六个药效团特征的药效团模型,并使用效力和选择性验证方法进行了验证。在效力验证期间,计算Guner-Henry分数以检查所生成模型的准确性,而在选择性验证中,评估能够识别选择性JAK1抑制剂的药效团模型。基于验证结果,选择了最佳药效团模型ADHRRR、DDHRRR、DDRRR、DPRRR、DHRRR、ADRRR、DDHRR和ADPRR,并针对Maybridge、Asinex、Chemdiv、Enamine、Lifechemicals和Zinc数据库进行虚拟筛选,以识别能够与JAK1结合的具有新型骨架的新分子。通过筛选共鉴定出4265个命中化合物,并检查其是否具有可接受的类药性质。经过ADME预测后,共选择了2856个命中化合物,并进行Glide分子对接以评估先导候选物的准确结合模式。基于与JAK1重要残基的结合能和氢键相互作用,筛选出90个分子。通过使用MM-GBSA计算和诱导契合对接方法计算蛋白质-配体复合物的结合自由能,对接结果得到了验证。随后,采用交叉对接方法识别选择性JAK1先导化合物。最后,选择了对JAK1具有强效和选择性的前五种先导化合物,并使用分子动力学模拟进行验证。此外,还对所选先导化合物进行了密度泛函理论研究。通过各种计算研究,我们观察到与药物鲁索替尼相比,这些先导化合物具有良好的效力和选择性。发现化合物T5923555和T5923531是最佳的,可以使用和方法进一步验证。