Center for Basic Medical Research & Department of Cardiovascular Surgery, TEDA International Cardiovascular Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin 300457, China.
The Institute of Cardiovascular Diseases, Tianjin University, Tianjin 300457, China.
Acta Biochim Biophys Sin (Shanghai). 2022 Mar 25;54(3):388-399. doi: 10.3724/abbs.2022009.
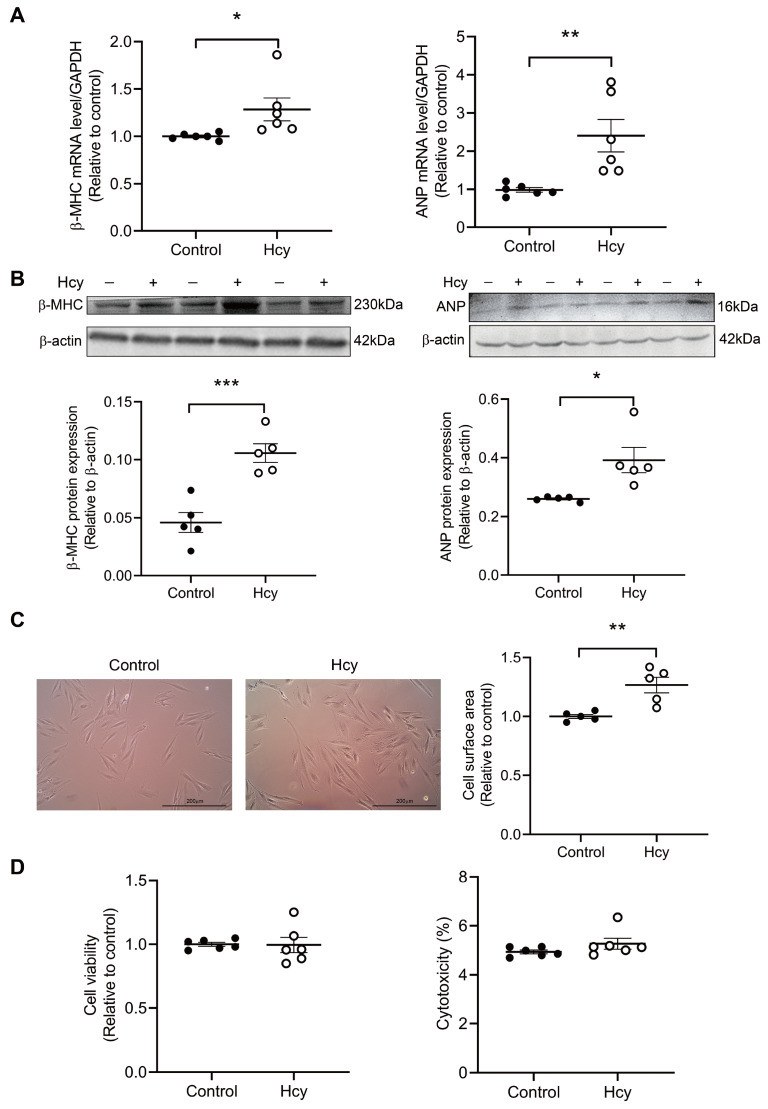
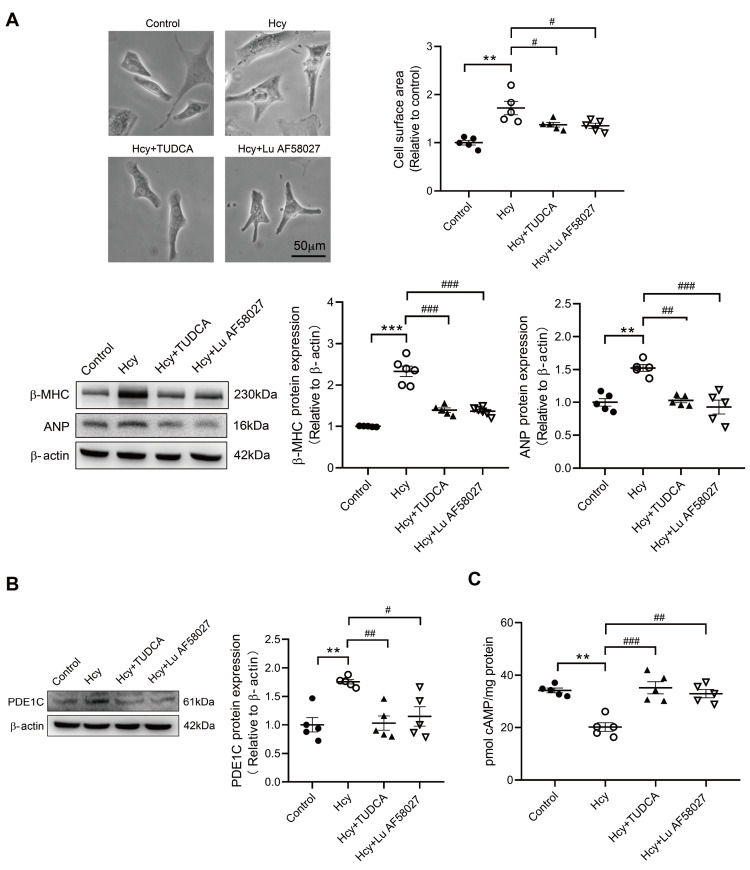
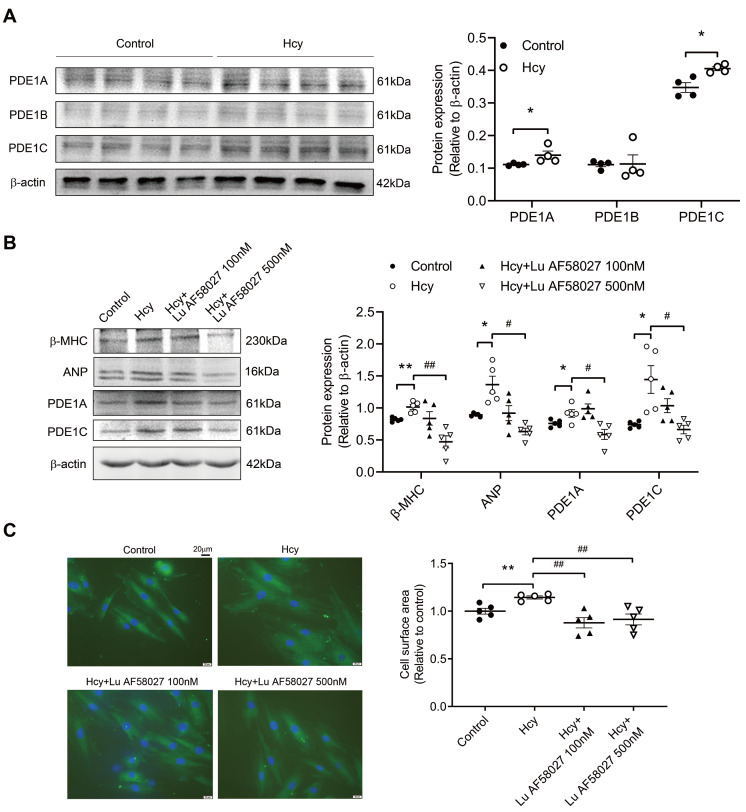
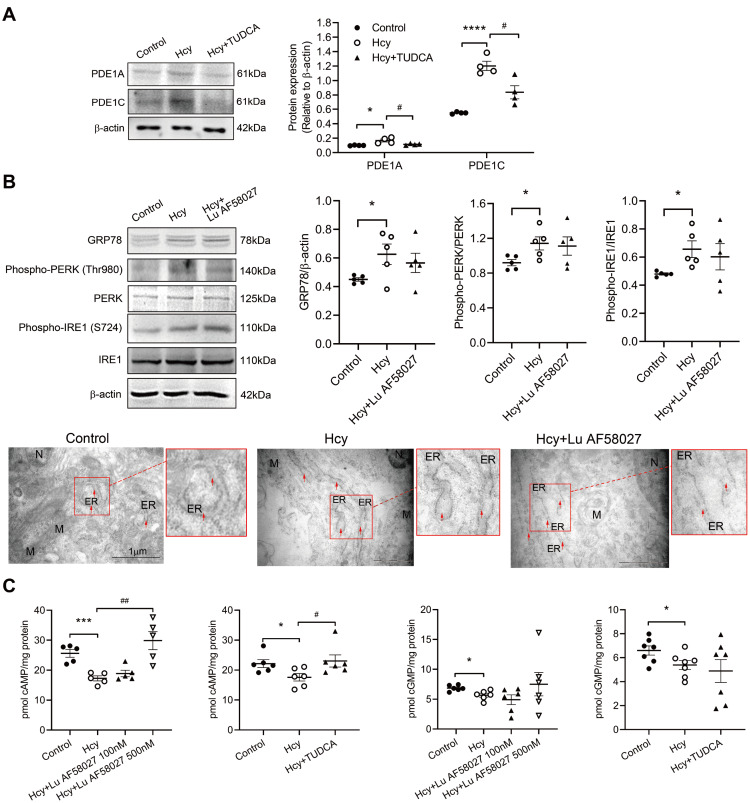
Although the association of elevated homocysteine level with cardiac hypertrophy has been reported, the molecular mechanisms by which homocysteine induces cardiac hypertrophy remain inadequately understood. In this study we aim to uncover the roles of cyclic nucleotide phosphodiesterase 1 (PDE1) and endoplasmic reticulum (ER) stress and their relationship to advance the mechanistic understanding of homocysteine-induced cardiac cell hypertrophy. H9c2 cells and primary neonatal rat cardiomyocytes are exposed to homocysteine with or without ER stress inhibitor TUDCA or PDE1-specific inhibitor Lu AF58027, or transfected with siRNAs targeting PDE1 isoforms prior to homocysteine-exposure. Cell surface area is measured and ultrastructure is examined by transmission electron microscopy. Hypertrophic markers, PDE1 isoforms, and ER stress molecules are detected by q-PCR and western blot analysis. Intracellular cGMP and cAMP are measured by ELISA. The results show that homocysteine causes the enlargement of H9c2 cells, increases the expressions of hypertrophic markers β-MHC and ANP, upregulates PDE1A and PDE1C, promotes the expressions of ER stress molecules, and causes ER dilatation and degranulation. TUDCA and Lu AF58027 downregulate β-MHC and ANP, and alleviate cell enlargement. TUDCA decreases PDE1A and PDE1C levels. Silencing of PDE1C inhibits homocysteine-induced hypertrophy, whereas PDE1A knockdown has minor effect. Both cAMP and cGMP are decreased after homocysteine-exposure, while only cAMP is restored by Lu AF58027 and TUDCA. TUDCA and Lu AF58027 also inhibit cell enlargement, downregulate ANP, β-MHC and PDE1C, and enhance cAMP level in homocysteine-exposed primary cardiomyocytes. ER stress mediates homocysteine-induced hypertrophy of cardiac cells via upregulating PDE1C expression Cyclic nucleotide, especially cAMP, is the downstream mediator of the ER stress-PDE1C signaling axis in homocysteine-induced cell hypertrophy.
尽管已经报道了高同型半胱氨酸水平与心脏肥大之间的关联,但同型半胱氨酸诱导心脏肥大的分子机制仍未得到充分理解。在这项研究中,我们旨在揭示环核苷酸磷酸二酯酶 1(PDE1)和内质网(ER)应激的作用及其与同型半胱氨酸诱导的心肌细胞肥大的关系,以深入了解其机制。用同型半胱氨酸处理 H9c2 细胞和原代新生大鼠心肌细胞,或用 ER 应激抑制剂 TUDCA 或 PDE1 特异性抑制剂 Lu AF58027 预处理,或用靶向 PDE1 同工型的 siRNA 转染后再用同型半胱氨酸处理。通过透射电子显微镜测量细胞表面积并检查超微结构。通过 q-PCR 和 Western blot 分析检测肥大标志物、PDE1 同工型和 ER 应激分子。通过 ELISA 测量细胞内 cGMP 和 cAMP。结果表明,同型半胱氨酸导致 H9c2 细胞增大,增加了β-MHC 和 ANP 的表达,上调了 PDE1A 和 PDE1C,促进了 ER 应激分子的表达,并导致 ER 扩张和脱颗粒。TUDCA 和 Lu AF58027 下调β-MHC 和 ANP,并减轻细胞增大。TUDCA 降低了 PDE1A 和 PDE1C 水平。沉默 PDE1C 可抑制同型半胱氨酸诱导的肥大,而 PDE1A 敲低的影响较小。同型半胱氨酸处理后,cAMP 和 cGMP 均减少,而 Lu AF58027 和 TUDCA 仅恢复 cAMP。TUDCA 和 Lu AF58027 还抑制细胞增大,下调 ANP、β-MHC 和 PDE1C,并增强同型半胱氨酸暴露的原代心肌细胞中的 cAMP 水平。ER 应激通过上调 PDE1C 表达介导同型半胱氨酸诱导的心肌细胞肥大。环核苷酸,特别是 cAMP,是 ER 应激-PDE1C 信号轴在同型半胱氨酸诱导细胞肥大中的下游介质。