Kalinowska-Tłuścik Justyna, Staroń Jakub, Krawczuk Anna, Mordalski Stefan, Warszycki Dawid, Satała Grzegorz, Hogendorf Adam S, Bojarski Andrzej J
Department of Crystal Chemistry and Crystal Physic, Faculty of Chemistry, Jagiellonian University Gronostajowa 2 30-387 Kraków Poland
Department of Medicinal Chemistry, Institute of Pharmacology Polish Academy of Sciences Smętna 12 31-343 Kraków Poland.
RSC Adv. 2018 May 22;8(33):18672-18681. doi: 10.1039/c8ra03107j. eCollection 2018 May 17.
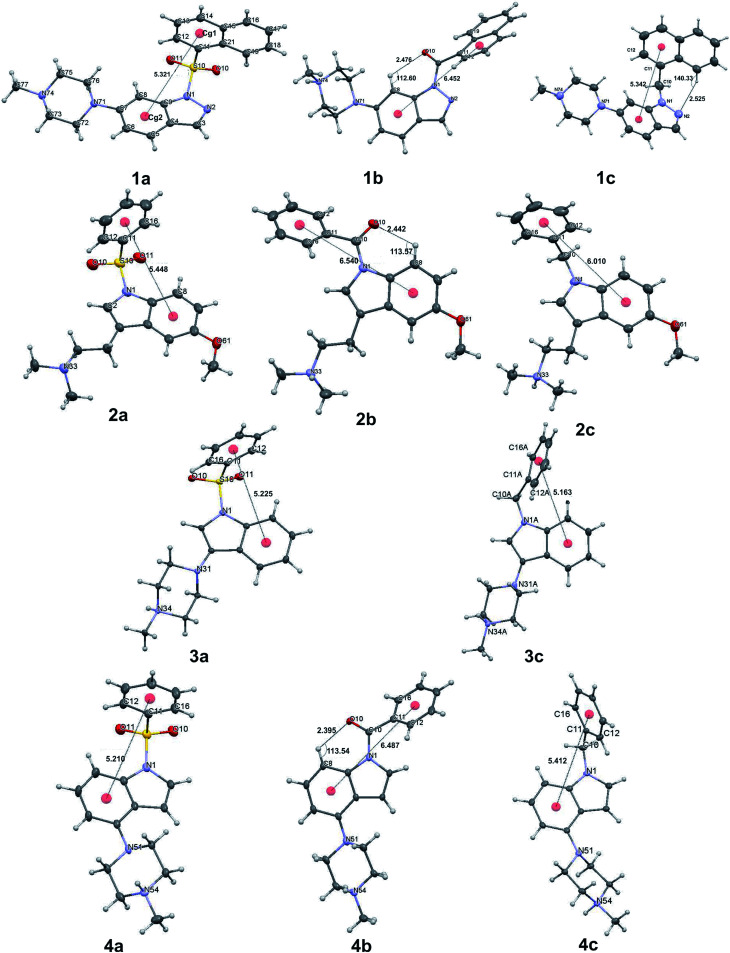
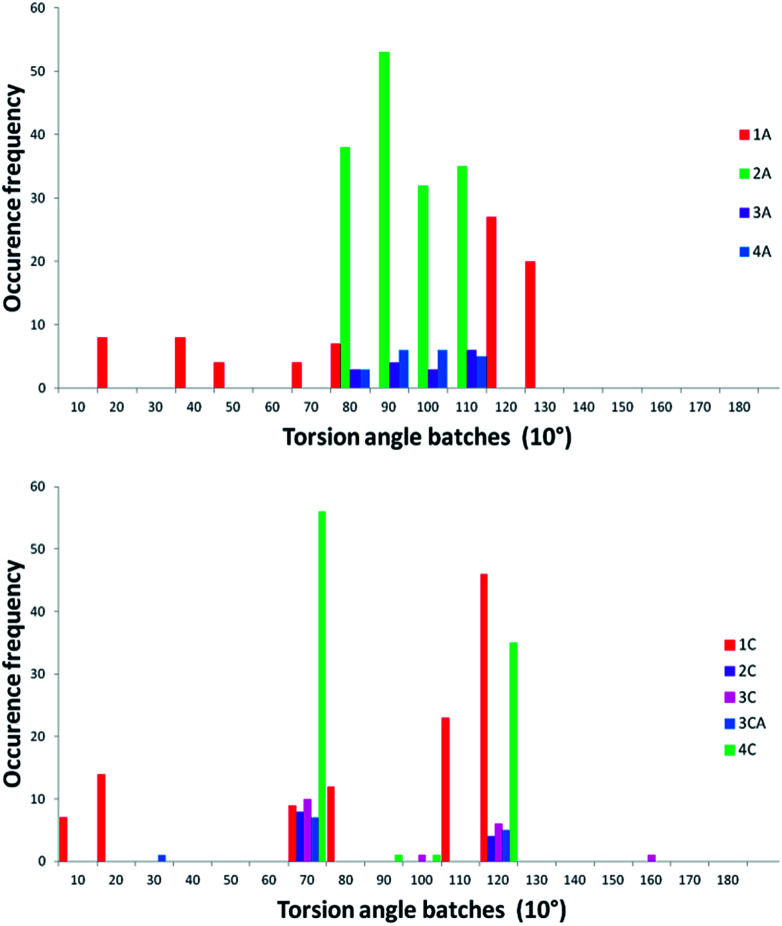
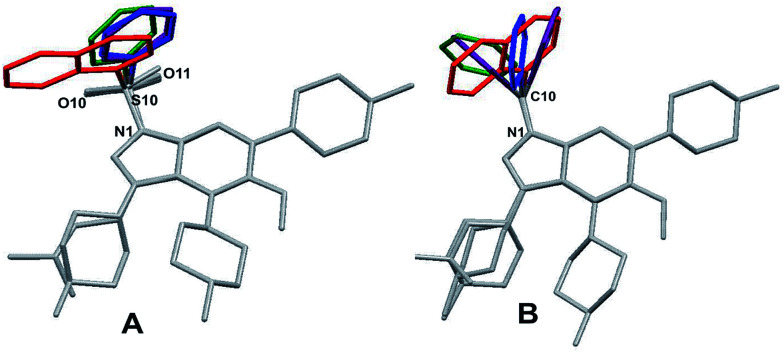
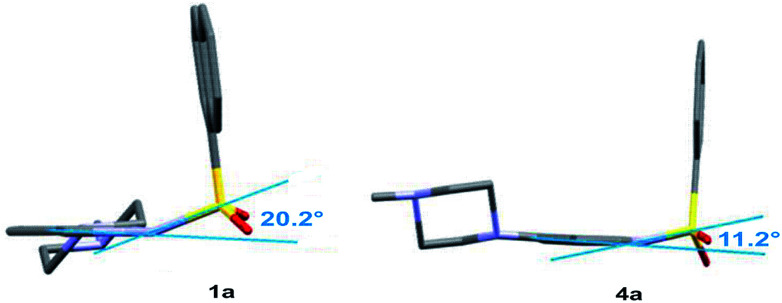
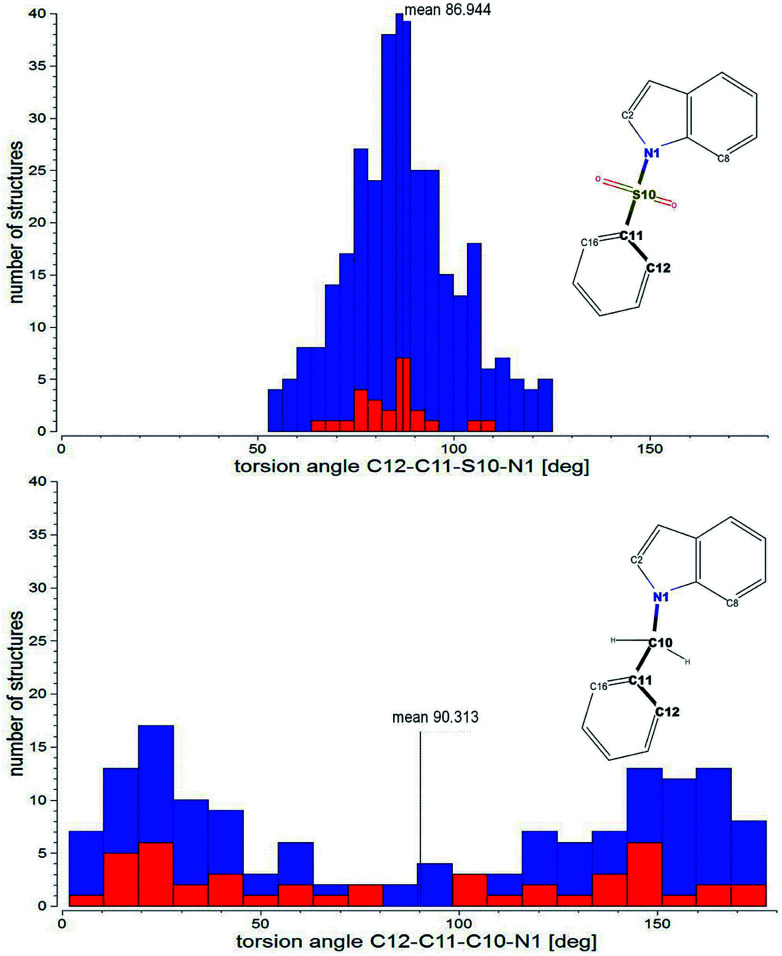
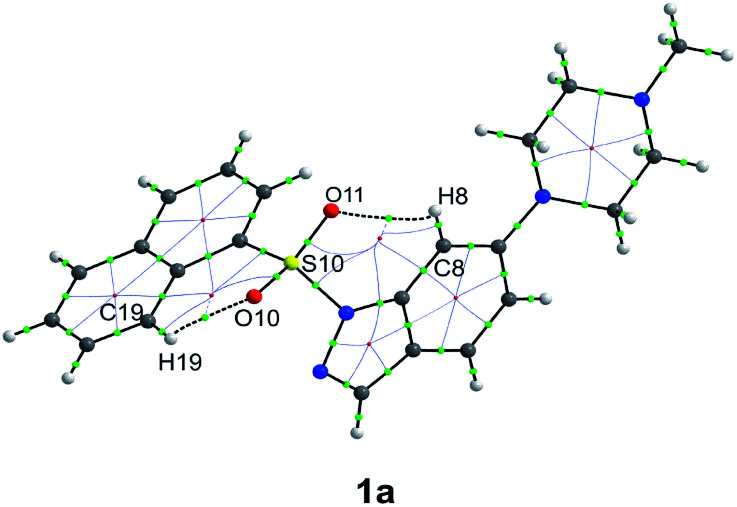
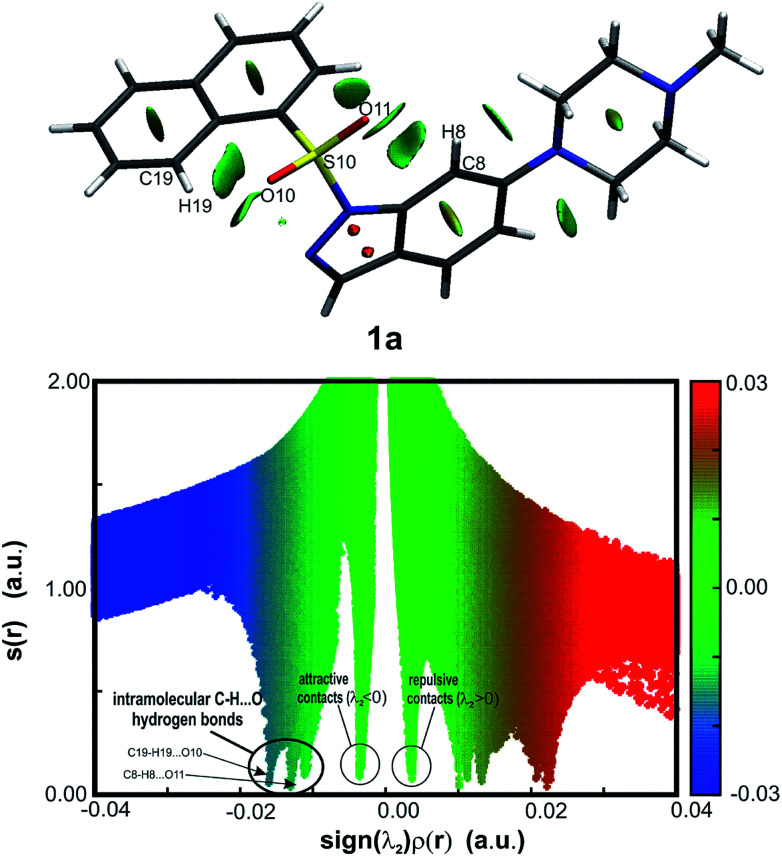
The development of compounds with enhanced activity and selectivity by a conserved spatial orientation of the pharmacophore elements has a long history in medicinal chemistry. Rigidified compounds are an example of this concept. However, the intramolecular interactions were seldom used as a basis for conformational restraints. Here, we show the weak intramolecular interactions that contribute to the relatively well-conserved geometry of N1-arylsulfonyl indole derivatives. The structure analysis along with quantum mechanics calculations revealed a crucial impact of the sulfonyl group on the compound geometry. The weak intramolecular C-H⋯O interaction stabilizes the mutual "facing" orientation of two aromatic fragments. These findings extend the pharmacological interpretation of the sulfonyl group role from the double hydrogen bond acceptor to the conformational scaffold based on intramolecular forces. This feature has, to date, been omitted in drug discovery. Our results should increase the awareness of researchers to consider the conformational preference when designing new compounds or improving computational methods.
通过药效基团元素的保守空间取向来开发具有增强活性和选择性的化合物,在药物化学领域有着悠久的历史。刚性化化合物就是这一概念的一个例子。然而,分子内相互作用很少被用作构象限制的基础。在此,我们展示了有助于N1-芳基磺酰基吲哚衍生物相对保守几何结构的弱分子内相互作用。结构分析以及量子力学计算揭示了磺酰基对化合物几何结构的关键影响。弱分子内C-H⋯O相互作用稳定了两个芳香片段的相互“面对”取向。这些发现将磺酰基作用的药理学解释从双氢键受体扩展到基于分子内力的构象支架。迄今为止,这一特征在药物发现中一直被忽视。我们的结果应提高研究人员在设计新化合物或改进计算方法时考虑构象偏好的意识。