Zhai Diandian, Wang Jinpeng, Hao Lina, Ma Congming, Ma Peng, Pan Yong, Jiang Juncheng
College of Safety Science and Engineering, Nanjing Tech University Nanjing Jiangsu CN 211800 China
RSC Adv. 2019 Nov 19;9(65):37747-37758. doi: 10.1039/c9ra07087g.
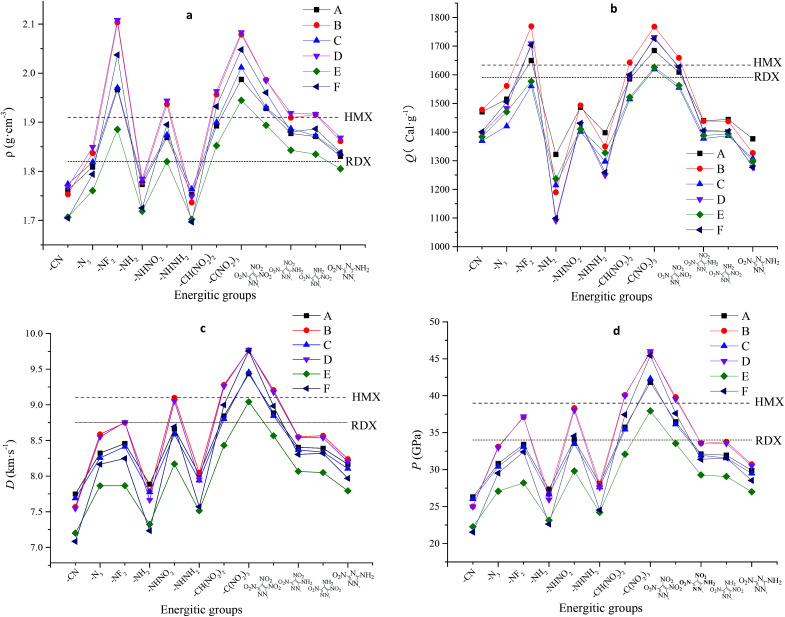
A series of bridged pyridine-based energetic derivatives were designed and their geometrical structures, electronic structures, heats of formation, detonation properties, thermal stabilities, thermodynamic properties and electrostatic potential were fully investigated using density functional theory. The results show that the steric hindrance effect is a decisive factor for structural stability, and the formation of intramolecular or intermolecular hydrogen bonds doesn't provide advantages to stabilize molecular structure, which was demonstrated by insertion of 3,4,5-trinitro-1-pyrazole, 3,4-dinitro-1-pyrazol-5-amine, 3,5-dinitro-1-pyrazol-4-amine and 3-nitro-1-1,2,4-triazol-5-amine. The azide group and azo bridge play an important role in improving the heats of formation of energetic pyridine-based materials. All designed molecules were found to have values of density ranging from 1.70 g cm (E6, F6) to 2.11 g cm (D3), values of detonation velocity ranging from 7.1 km s (F1) to 9.77 km s (D8), and values of detonation pressure ranging from 21.5 GPa (F1) to 46.0 GPa (D8). When a p-π conjugation formed between the nitrogen atom and pyridine ring, the bond between nitrogen and hydrogen atoms may be broken as the trigger bond.
设计了一系列桥连吡啶基含能衍生物,并使用密度泛函理论对其几何结构、电子结构、生成热、爆轰性能、热稳定性、热力学性质和静电势进行了全面研究。结果表明,空间位阻效应是结构稳定性的决定性因素,分子内或分子间氢键的形成对稳定分子结构并无优势,这通过插入3,4,5-三硝基-1-吡唑、3,4-二硝基-1-吡唑-5-胺、3,5-二硝基-1-吡唑-4-胺和3-硝基-1,2,4-三唑-5-胺得以证明。叠氮基和偶氮桥在提高吡啶基含能材料的生成热方面起着重要作用。发现所有设计分子的密度值范围为1.70 g/cm³(E6,F6)至2.11 g/cm³(D3),爆速值范围为7.1 km/s(F1)至9.77 km/s(D8),爆压值范围为21.5 GPa(F1)至46.0 GPa(D8)。当氮原子与吡啶环之间形成p-π共轭时,氮与氢原子之间的键可能会作为引发键断裂。