Nazir Majid, Abbasi Muhammad Athar, Siddiqui Sabahat Zahra, Raza Hussain, Hassan Mubashir, Ali Shah Syed Adnan, Shahid Muhammad, Seo Sung-Yum
Department of Chemistry, Government College University Lahore-54000 Pakistan
College of Natural Science, Department of Biological Sciences, Kongju National University Gongju 32588 South Korea.
RSC Adv. 2018 Jul 19;8(46):25920-25931. doi: 10.1039/c8ra04987d.
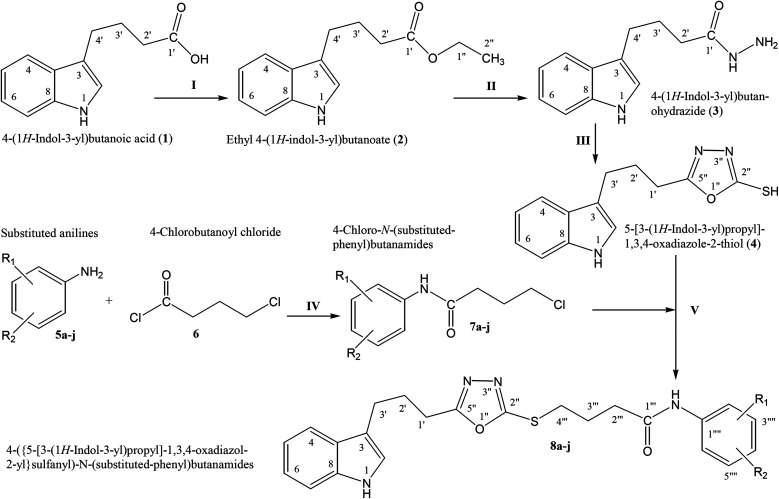
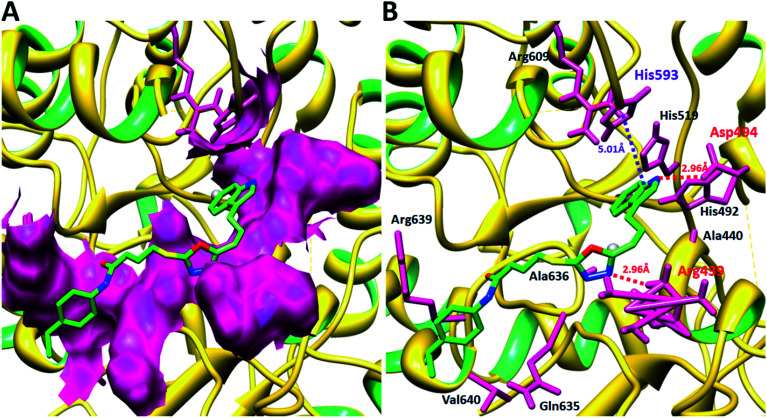
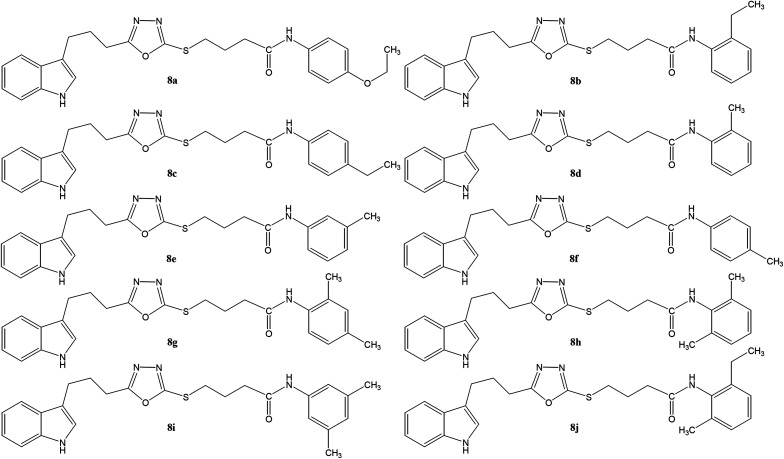
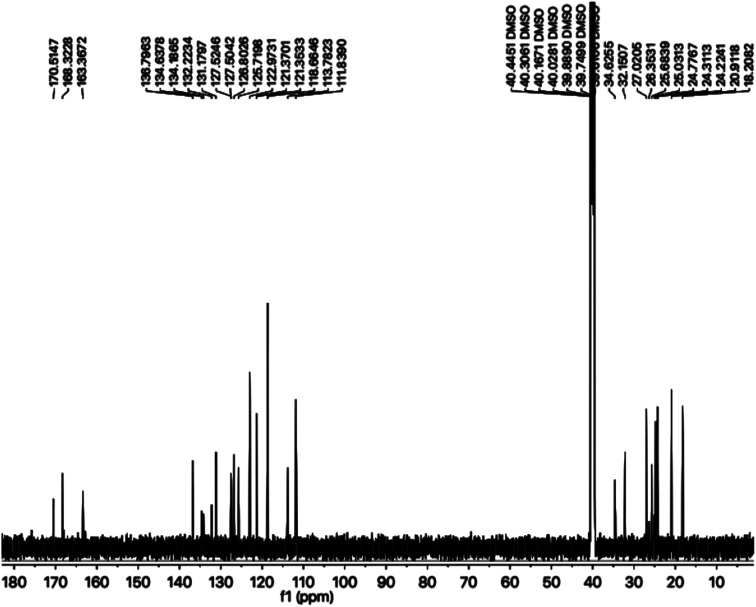
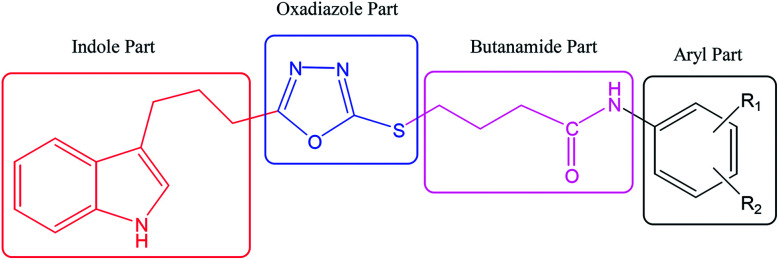
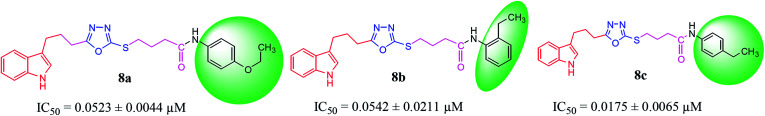
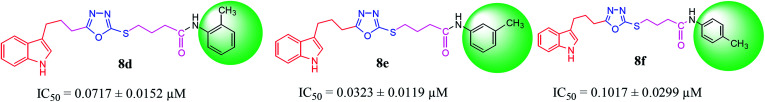
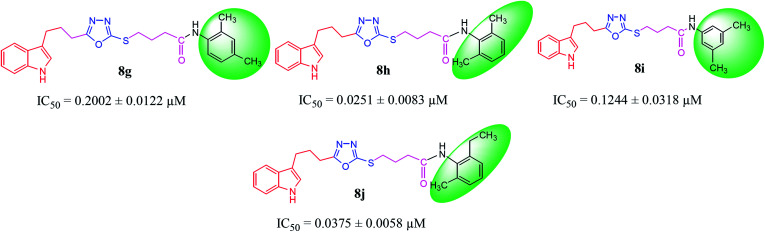
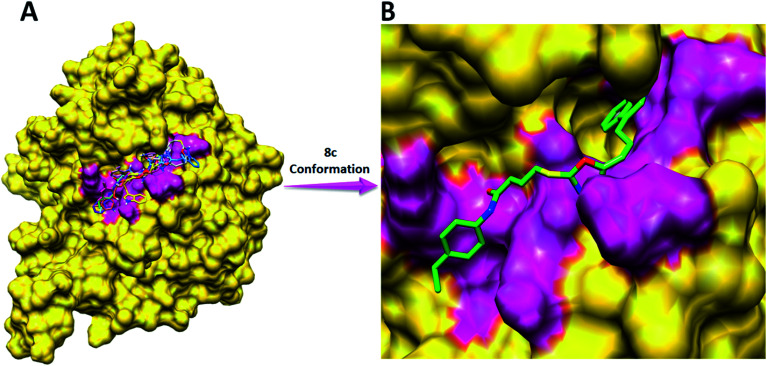
In the study presented herein, 4-(1-indol-3-yl)butanoic acid (1) was sequentially transformed in the first phase into ethyl 4-(1-indol-3-yl)butanoate (2), 4-(1-indol-3-yl)butanohydrazide (3) and 5-[3-(1-indol-3-yl)propyl]-1,3,4-oxadiazole-2-thiol (4) as a nucleophile. In the second phase, various electrophiles were synthesized by reacting substituted-anilines, 5a-j, with 4-chlorobutanoyl chloride (6) to afford 4-chloro--(substituted-phenyl)butanamides (7a-j). In the final phase, nucleophilic substitution reaction of 4 was carried out with different electrophiles, 7a-j, to achieve novel indole based oxadiazole scaffolds with -(substituted-phenyl)butamides (8a-j). The structural confirmation of all the as-synthesized compounds was performed by spectral and elemental analysis. These molecules were screened for their inhibitory potential against urease enzyme and were found to be potent inhibitors. The results of enzyme inhibitory kinetics showed that compound 8c inhibited the enzyme competitively with a value 0.003 μM. The results of the study of these scaffolds were in full agreement with the experimental data and the ligands showed good binding energy values. The hemolytic study revealed their mild cytotoxicity towards cell membranes and hence, these molecules can be regarded as valuable therapeutic agents in drug designing programs.
在本文所展示的研究中,4-(1-吲哚-3-基)丁酸(1)在第一阶段依次转化为4-(1-吲哚-3-基)丁酸乙酯(2)、4-(1-吲哚-3-基)丁酰肼(3)和5-[3-(1-吲哚-3-基)丙基]-1,3,4-恶二唑-2-硫醇(4)作为亲核试剂。在第二阶段,通过使取代苯胺5a-j与4-氯丁酰氯(6)反应合成了各种亲电试剂,得到4-氯-(取代苯基)丁酰胺(7a-j)。在最后阶段,使4与不同的亲电试剂7a-j进行亲核取代反应,以获得具有-(取代苯基)丁酰胺的新型基于吲哚的恶二唑支架(8a-j)。通过光谱和元素分析对所有合成化合物进行了结构确认。对这些分子进行了脲酶抑制潜力的筛选,发现它们是有效的抑制剂。酶抑制动力学结果表明,化合物8c以0.003 μM的Ki值竞争性抑制该酶。对这些支架的研究结果与实验数据完全一致,并且这些配体显示出良好的结合能值。溶血研究揭示了它们对细胞膜的轻度细胞毒性,因此,这些分子可被视为药物设计项目中有价值的治疗剂。