University of Trieste, Piazzale Europa, 1, 34127, Trieste, Italy.
Institute for Maternal and Child Health - IRCCS Burlo Garofolo, Trieste, Italy.
Ital J Pediatr. 2022 May 12;48(1):72. doi: 10.1186/s13052-022-01267-w.
Wolf-Hirschhorn syndrome (WHS) is a well-defined disorder, whose core phenotype encompasses growth restriction, facial gestalt, intellectual disability and seizures. Nevertheless, great phenotypic variability exists due to the variable extent of the responsible 4p deletion. In addition, exome sequencing analyses, recently identified two genes, namely NSD2 and NELFA, whose loss-of-function variants contribute to a clinical spectrum consistent with atypical or partial WHS. The observation of patients exhibiting clinical features resembling WHS, with only mild developmental delay and without the typical dysmorphic features, carrying microdeletions sparing NSD2, has lead to the hypothesis that NSD2 is responsible for the intellectual disability and the facial gestalt of WHS. While presenting some of the typical findings of WHS (intellectual disability, facial gestalt, microcephaly, growth restriction and congenital heart defects), NSD2-deleted children tend to display a milder spectrum of skeletal abnormalities, usually consisting of clinodactyly, and do not exhibit seizures. We describe the clinical picture of a child with WHS due to a de novo mutation of NSD2 and discuss the clinical and diagnostic implications.
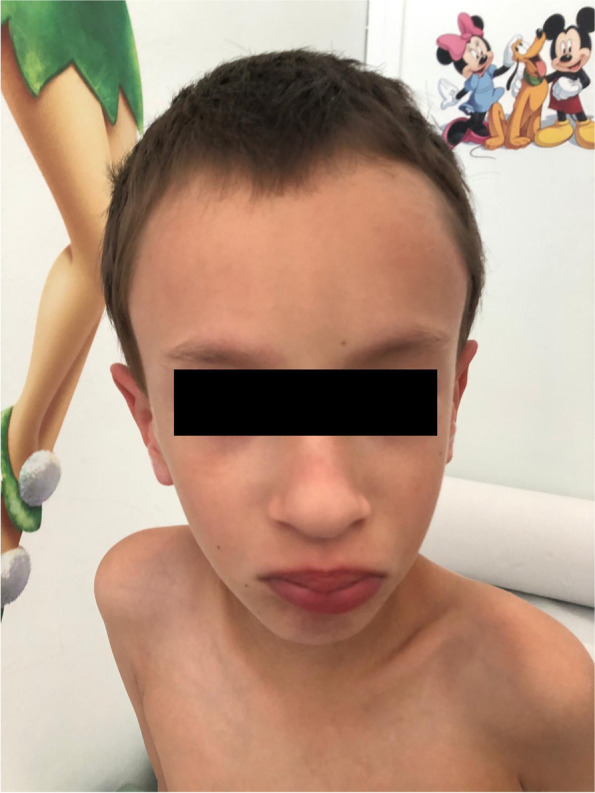
A 6-year-old boy was evaluated for a history of intrauterine growth restriction, low birth weight, neonatal hypotonia, and psychomotor delay. No episodes of seizure were reported. At physical examination, he displayed marphanoid habitus, muscle hypotrophy and facial dysmorphisms consisting in high frontal hairline, upslanting palpebral fissures and full lips with bifid ugula. Cryptorchidism, shawl scrotum, mild clinodactyly of the right little finger and bilateral syndactyly of the II and III toes with sandal gap were also noted. The radiographic essay demonstrated delayed bone age and echocardiography showed mild mitral prolapse. Whole genome sequencing analysis revealed a heterozygous de novo variant of NSD2 (c.2523delG).
Full WHS phenotype likely arises from the cumulative effect of the combined haploinsufficiency of several causative genes mapping within the 4p16.3 region, as a contiguous genes syndrome, with slightly different phenotypes depending on the specific genes involved in the deletion. When evaluating children with pictures resembling WHS, in absence of seizures, clinicians should consider this differential diagnosis.
沃尔夫-赫希霍恩综合征(WHS)是一种明确的疾病,其核心表型包括生长受限、面部特征、智力障碍和癫痫。然而,由于负责的 4p 缺失程度不同,存在很大的表型变异性。此外,外显子组测序分析最近确定了两个基因,即 NSD2 和 NELFA,其功能丧失变异导致与非典型或部分 WHS 一致的临床谱。观察到表现出类似于 WHS 的临床特征的患者,只有轻度发育迟缓,没有典型的畸形特征,携带 NSD2 缺失的微缺失,导致了 NSD2 负责 WHS 的智力障碍和面部特征的假说。虽然表现出 WHS 的一些典型发现(智力障碍、面部特征、小头畸形、生长受限和先天性心脏缺陷),但 NSD2 缺失的儿童往往表现出较轻的骨骼异常谱,通常包括指骨弯曲,并且没有癫痫发作。我们描述了一个由于 NSD2 从头突变导致的 WHS 儿童的临床图片,并讨论了临床和诊断意义。
一名 6 岁男孩因宫内生长受限、低出生体重、新生儿低张力和精神运动发育迟缓而接受评估。没有癫痫发作的报道。体格检查显示马凡体型、肌肉萎缩和面部畸形,包括高额发际线、上睑裂和丰满的嘴唇,分裂的悬雍垂。隐睾、披肩阴囊、右小指轻度弯曲和 II 和 III 脚趾双侧并指,伴有凉鞋间隙。影像学检查显示骨龄延迟,超声心动图显示轻度二尖瓣脱垂。全基因组测序分析显示 NSD2(c.2523delG)杂合性从头变异。
完整的 WHS 表型可能是由于几个位于 4p16.3 区域内的致病基因的联合杂合功能缺失引起的,作为一个连续的基因综合征,由于涉及缺失的特定基因略有不同,表型也略有不同。当评估具有类似于 WHS 的图片的儿童时,在没有癫痫发作的情况下,临床医生应考虑这种鉴别诊断。