Pharmacokinetic Research Laboratories, Ono Pharmaceutical Co., Ltd., Ibaraki, Japan.
Drug Metabolism and Pharmacokinetics, In Vitro In Vivo Translation, GlaxoSmithKline R&D, Stevenage, UK.
CPT Pharmacometrics Syst Pharmacol. 2022 Jul;11(7):919-933. doi: 10.1002/psp4.12807. Epub 2022 Jun 6.
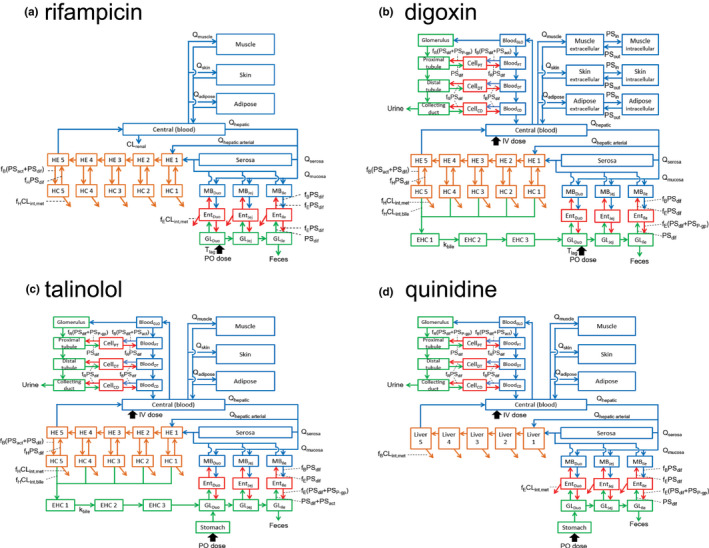
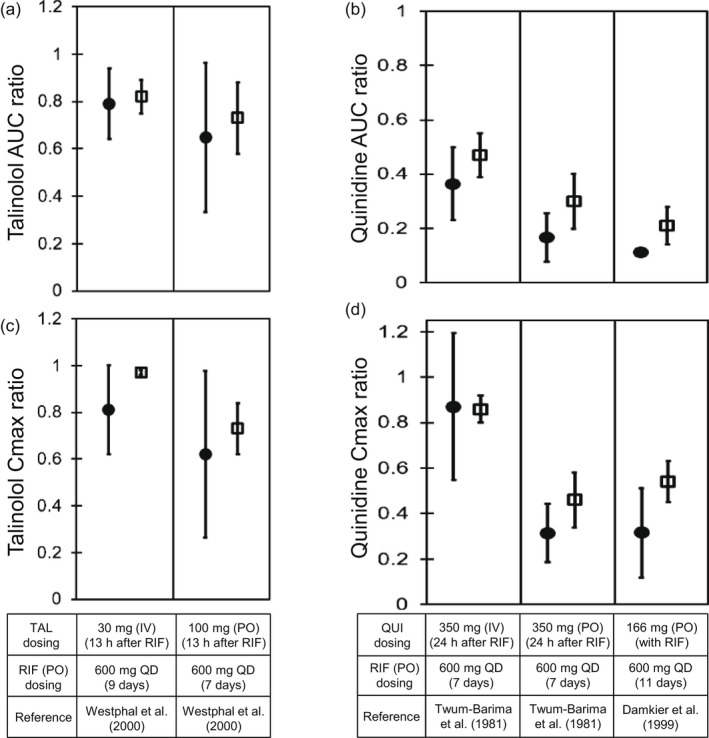
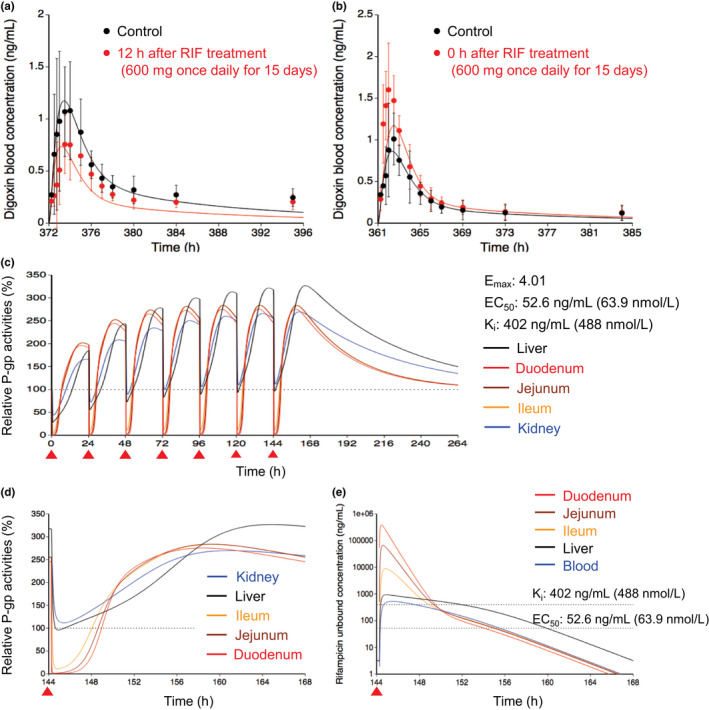
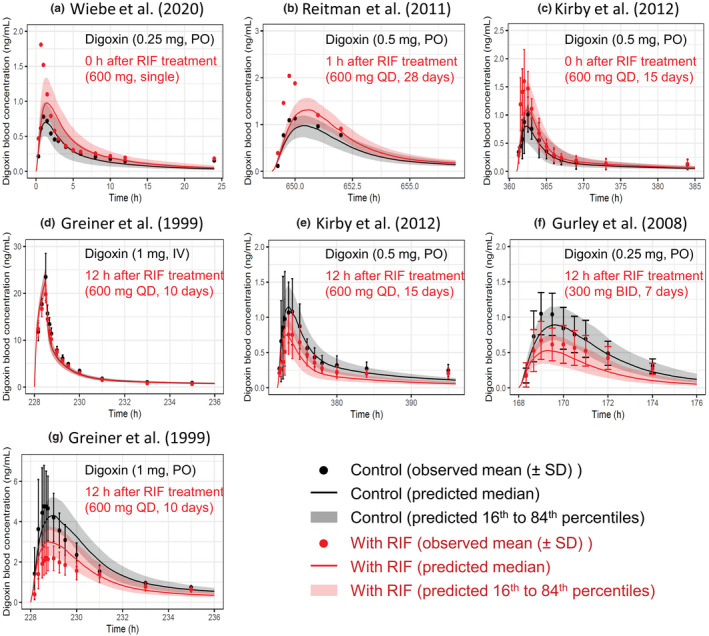
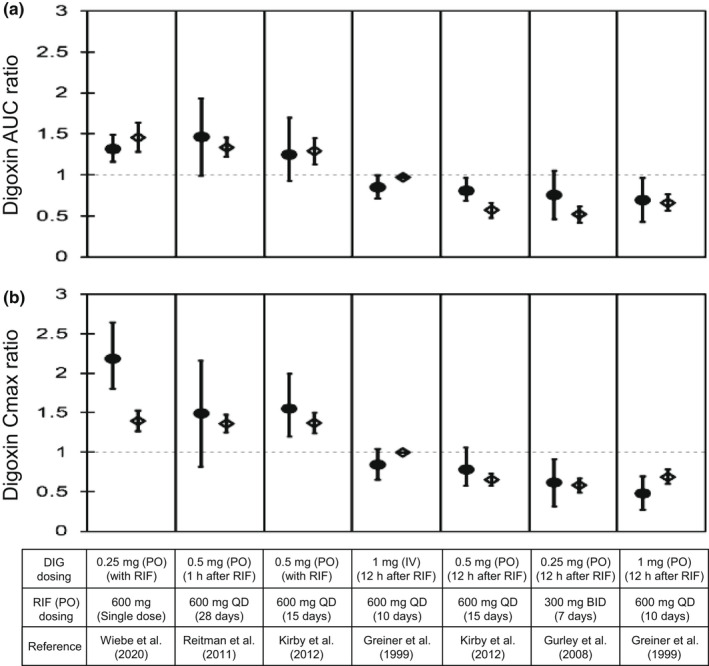
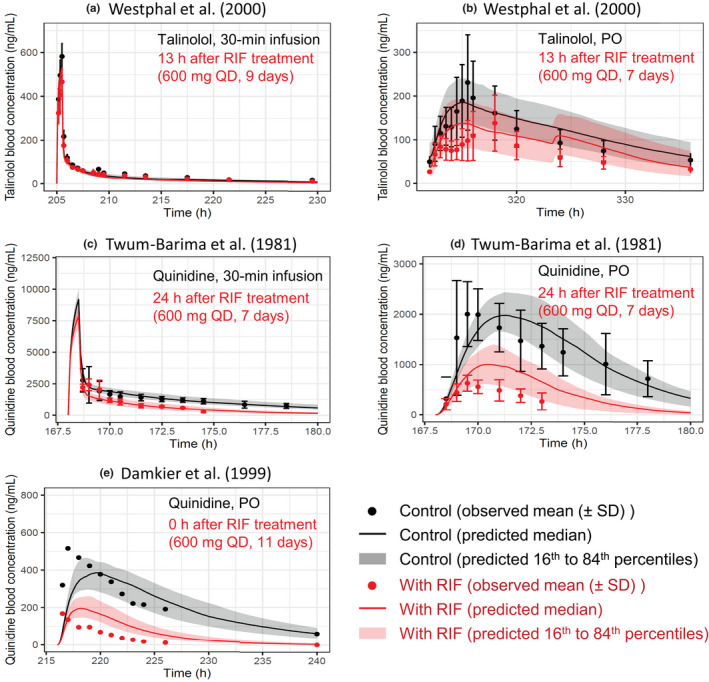
P-glycoprotein (P-gp) is an efflux transporter that plays an important role in the pharmacokinetics of its substrate, and P-gp activities can be altered by induction and inhibition effects of rifampicin. This study aimed to establish a physiologically based pharmacokinetic (PBPK) model of rifampicin to predict the P-gp-mediated drug-drug interactions (DDIs) and assess the DDI impact in the intestine, liver, and kidney. The induction and inhibition parameters of rifampicin for P-gp were estimated using two of seven DDI cases of rifampicin and digoxin and incorporated into our previously constructed PBPK model of rifampicin. The constructed rifampicin model was verified using the remaining five DDI cases with digoxin and five DDI cases with other P-gp substrates (talinolol and quinidine). Based on the established PBPK model, following repeated dosing of 600 mg rifampicin, the deduced net effect was an approximately threefold induction in P-gp activities in the intestine, liver, and kidney. Furthermore, in all 12 cases the predicted area under the plasma concentration-time curve ratios of the P-gp substrates were within the predefined acceptance criteria with various dosing regimens. Intestinal effects of P-gp-mediated DDIs had their greatest impact on the pharmacokinetics of digoxin and talinolol, with a minimal impact on the liver and kidney. For quinidine, predicted intestinal P-gp/cytochrome P450 3A-mediated DDIs were slightly underestimated because of the complexity of nonlinearity and transporter-enzyme interplay. These findings demonstrate that our rifampicin model can be applicable to quantitatively predict the net impact of P-gp induction and/or inhibition on diverse P-gp substrates and investigate the magnitude of DDIs in each tissue.
P-糖蛋白(P-gp)是一种外排转运体,在其底物的药代动力学中起着重要作用,并且 P-gp 的活性可以通过利福平的诱导和抑制作用改变。本研究旨在建立利福平的基于生理学的药代动力学(PBPK)模型,以预测 P-gp 介导的药物相互作用(DDI),并评估在肠道、肝脏和肾脏中的 DDI 影响。使用利福平与地高辛的七种 DDI 情况中的两种来估计利福平对 P-gp 的诱导和抑制参数,并将其纳入我们之前构建的利福平 PBPK 模型中。使用剩余的五种地高辛和五种其他 P-gp 底物(他林洛尔和奎尼丁)的 DDI 情况来验证构建的利福平模型。基于建立的 PBPK 模型,在重复给予 600mg 利福平后,推断出 P-gp 在肠道、肝脏和肾脏中的活性大约增加了三倍。此外,在所有 12 种情况下,根据不同的给药方案,预测的 P-gp 底物的血浆浓度-时间曲线下面积比均在预设的可接受标准范围内。P-gp 介导的 DDI 的肠道作用对地高辛和他林洛尔的药代动力学影响最大,对肝脏和肾脏的影响最小。对于奎尼丁,由于非线性和转运体-酶相互作用的复杂性,预测的肠道 P-gp/细胞色素 P450 3A 介导的 DDI 略低于实际情况。这些发现表明,我们的利福平模型可适用于定量预测 P-gp 诱导和/或抑制对各种 P-gp 底物的净影响,并研究每种组织中 DDI 的程度。