From the Department of Neurology (B.Z., C.Z., L.L., Y.S., Y.Z., C.Y.), Qilu Hospital (Qingdao), Cheeloo College of Medicine, Shandong University; Research Institute of Neuromuscular and Neurodegenerative Diseases and Department of Neurology (T.D., D.Z., F.L., C.Y.), Qilu Hospital, Shandong University; Department of Medicine Experimental Center (X.M.), Qilu Hospital (Qingdao), Cheeloo College of Medicine, Shandong University; Key Laboratory of the Ministry of Education for Medicinal Resources and Natural Pharmaceutical Chemistry (Y.Y.), College of Life Sciences, Shanxi Normal University, Xi'an, China; Division of Neuropathology (J.-Q.L.), Department of Pathology and Molecular Medicine, McMaster University, Hamilton, Ontario, Canada; Mitochondrial Medicine Laboratory (C.Y.), Qilu Hospital (Qingdao); and Brain Science Research Institute (C.Y.), Shandong University, Jinan, China.
Neurol Neuroimmunol Neuroinflamm. 2022 May 17;9(4). doi: 10.1212/NXI.0000000000001184. Print 2022 Jul.
Sporadic late-onset nemaline myopathy (SLONM) is a treatable or otherwise fatal myopathy. Diagnosis of SLONM is still challenging, and no therapeutic consensus has been achieved. Here, we reported the clinicopathologic features and long-term follow-up data of SLONM in a Chinese cohort.
We performed a retrospective evaluation of clinical, pathologic, and treatment outcomes of 17 patients with SLONM diagnosed between March 1986 and April 2021 at our neuromuscular center. Immunohistochemistry (IHC) with antibodies against 5 Z-disc-associated proteins was performed in the muscle biopsies of SLONM to identify a potential pathologic marker in aid of diagnosis. In comparison, we also performed muscle IHC in patients with selective type II fiber atrophy (n = 22), neurogenic atrophy (n = 22), mitochondrial myopathy (n = 5), immune-mediated necrotizing myopathy (n = 5), and normal controls (n = 5).
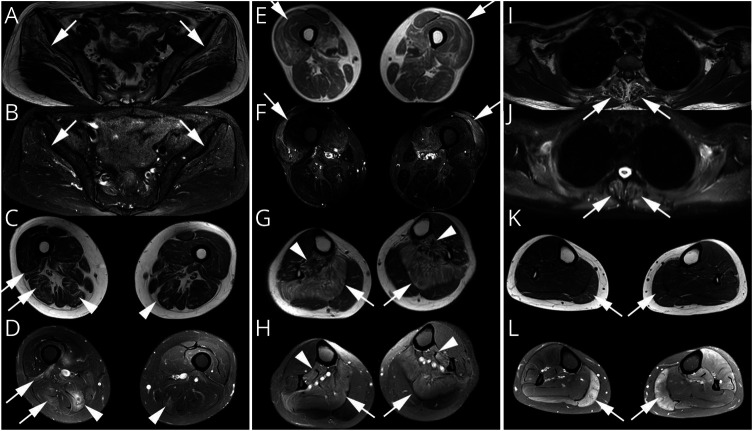
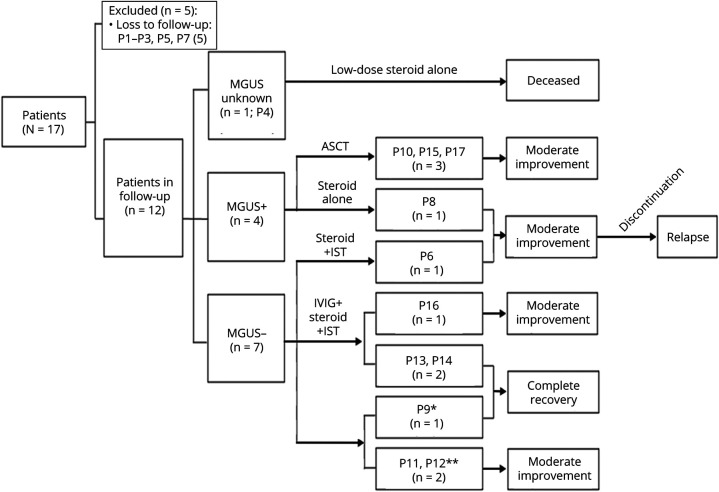
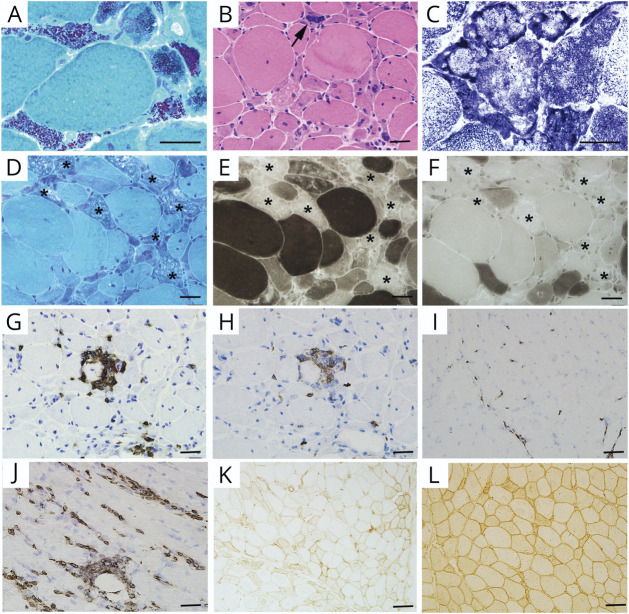
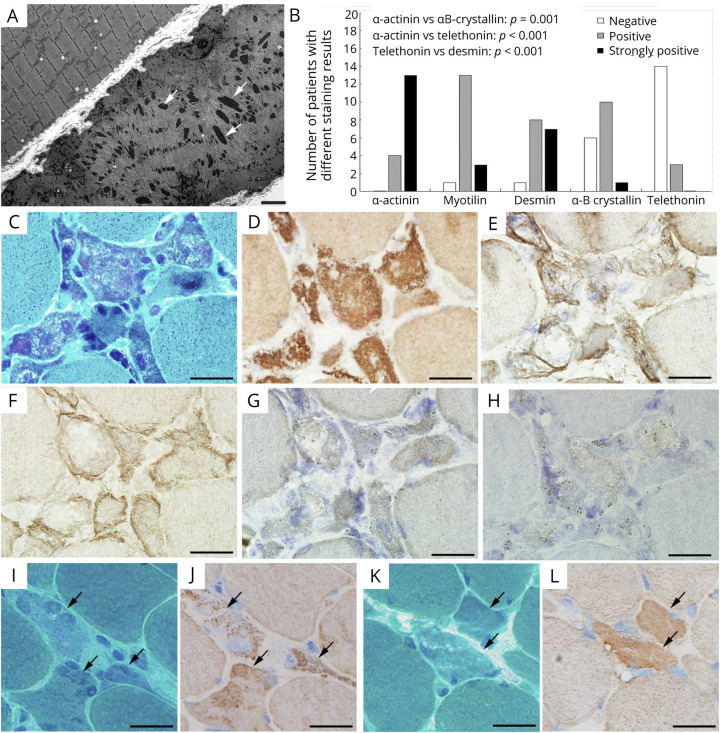
Most of the patients exhibited asymmetric limb muscles weakness (71%, 12/17) and neck extensor weakness (53%, 9/17). Immunofixation electrophoresis was performed in 11 patients, and 4 of them were identified with monoclonal gammopathy of undetermined significance (MGUS). EMG from 16 patients demonstrated a myopathic pattern with spontaneous activities in 69% (11/16) of them. Muscle MRI showed preferential involvement of paraspinal, gluteus minimus and medius, semimembranosus, and soleus muscles. Suspected nemaline bodies on modified Gomori trichrome were confirmed by IHC using anti-α-actinin antibody (100%, 17/17), anti-myotilin antibody (94%, 16/17), anti-desmin antibody (94%, 16/17), anti-α-B crystallin antibody (65%, 11/17), and anti-telethonin antibody (18%, 3/17) with various positive rates. Notably, anti-α-actinin IHC showed the highest percentage of strongly positive staining (77%, 13/17), being the only one without negative results. Moderate improvement following autologous stem cell transplantation (ASCT) was noted in 3/4 patients with MGUS; favorable outcomes were also achieved in 6/7 patients without MGUS, including 3 patients with complete recovery who were given a combined treatment of prednisone and another immunosuppressant.
SLONM is a treatable myopathy with ASCT or traditional immunotherapy, especially when combined with steroids and immunosuppressants. Anti-α-actinin immunostaining is the most reliable pathologic marker to identify rod-bearing fibers, and it should be performed routinely in adult patients with undiagnosed nonnecrotic myopathies.
散发性晚发性杆状体肌病(SLONM)是一种可治疗或致命的肌病。SLONM 的诊断仍然具有挑战性,并且尚未达成治疗共识。在此,我们报告了在中国队列中诊断的 17 例 SLONM 的临床病理特征和长期随访数据。
我们对 1986 年 3 月至 2021 年 4 月在我们的神经肌肉中心诊断的 17 例 SLONM 患者的临床、病理和治疗结果进行了回顾性评估。对 SLONM 的肌肉活检进行了 5 种 Z 盘相关蛋白的免疫组化(IHC)检测,以鉴定潜在的病理标志物以辅助诊断。相比之下,我们还对选择性 II 型纤维萎缩(n=22)、神经源性萎缩(n=22)、线粒体肌病(n=5)、免疫介导的坏死性肌病(n=5)和正常对照组(n=5)的患者进行了肌肉 IHC。
大多数患者表现为不对称性肢体肌肉无力(71%,12/17)和颈部伸肌无力(53%,9/17)。11 例患者进行了免疫固定电泳,其中 4 例被确定为意义未明的单克隆丙种球蛋白血症(MGUS)。16 例患者的肌电图显示肌病模式,其中 69%(11/16)有自发性活动。肌肉 MRI 显示脊柱旁、臀小肌和中肌、半膜肌和比目鱼肌优先受累。改良 Gomori 三色染色上可疑的杆状体用抗α-肌动蛋白抗体(100%,17/17)、抗肌球蛋白抗体(94%,16/17)、抗结蛋白抗体(94%,16/17)、抗α-B 晶体蛋白抗体(65%,11/17)和抗 telethonin 抗体(18%,3/17)进行免疫组化确认,阳性率各不相同。值得注意的是,抗α-肌动蛋白 IHC 显示出最高比例的强阳性染色(77%,13/17),是唯一没有阴性结果的染色。4 例 MGUS 患者中有 3 例接受自体干细胞移植(ASCT)后有中度改善;7 例无 MGUS 患者中也取得了良好的结果,包括 3 例完全恢复的患者,他们接受了泼尼松和另一种免疫抑制剂的联合治疗。
SLONM 是一种可治疗的肌病,可采用 ASCT 或传统免疫治疗,尤其是与皮质类固醇和免疫抑制剂联合使用时。抗α-肌动蛋白免疫染色是鉴定杆状纤维最可靠的病理标志物,应常规用于诊断不明的非坏死性肌病的成年患者。