Centre for Cancer Genetic Epidemiology, Department of Public Health and Primary Care, University of Cambridge, Cambridge, CB1 8RN, UK.
Department of Genetics and Computational Biology, QIMR Berghofer Medical Research Institute, Brisbane, QLD, 4006, Australia.
Genome Med. 2022 May 18;14(1):51. doi: 10.1186/s13073-022-01052-8.
Protein truncating variants in ATM, BRCA1, BRCA2, CHEK2, and PALB2 are associated with increased breast cancer risk, but risks associated with missense variants in these genes are uncertain.
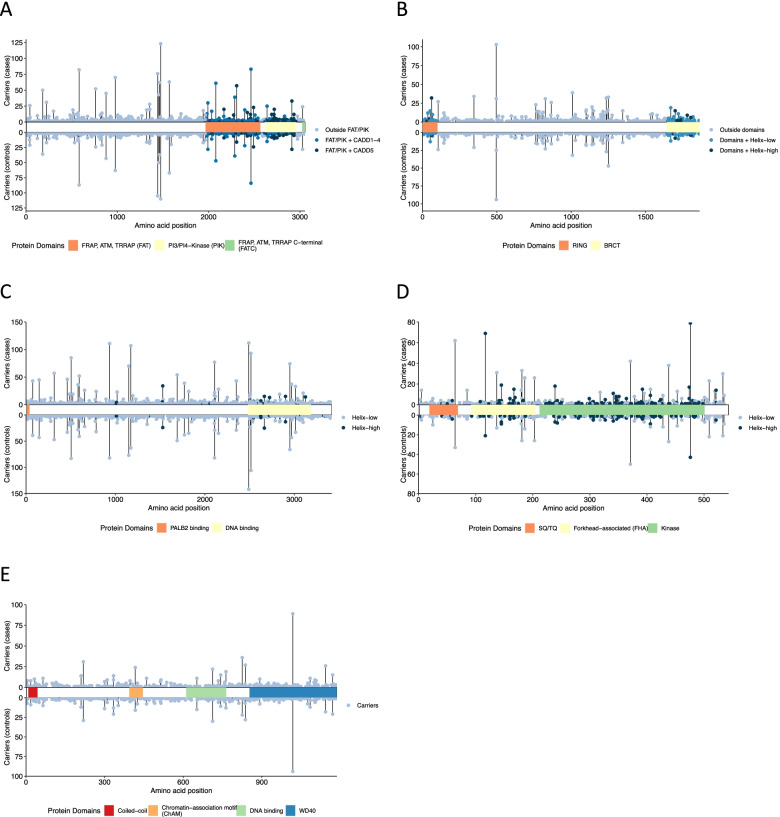
We analyzed data on 59,639 breast cancer cases and 53,165 controls from studies participating in the Breast Cancer Association Consortium BRIDGES project. We sampled training (80%) and validation (20%) sets to analyze rare missense variants in ATM (1146 training variants), BRCA1 (644), BRCA2 (1425), CHEK2 (325), and PALB2 (472). We evaluated breast cancer risks according to five in silico prediction-of-deleteriousness algorithms, functional protein domain, and frequency, using logistic regression models and also mixture models in which a subset of variants was assumed to be risk-associated.
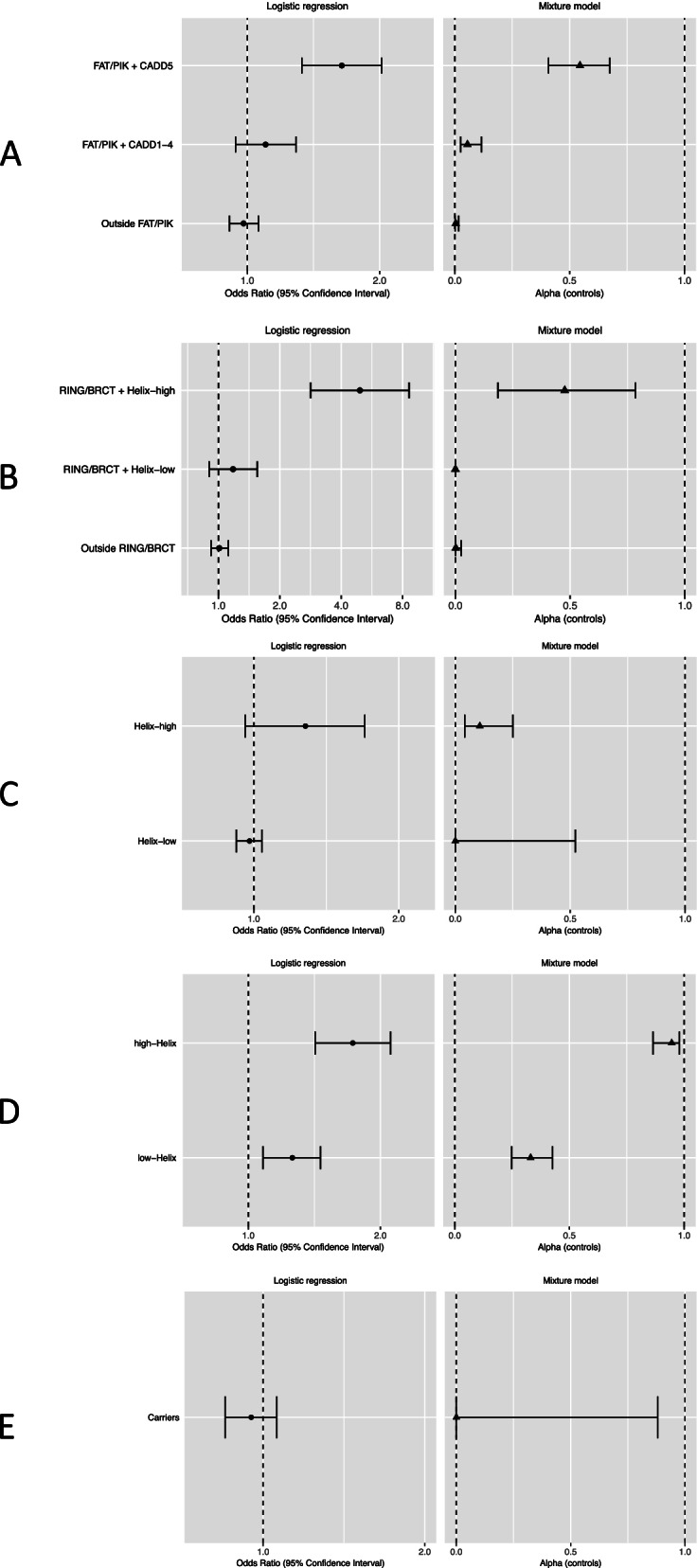
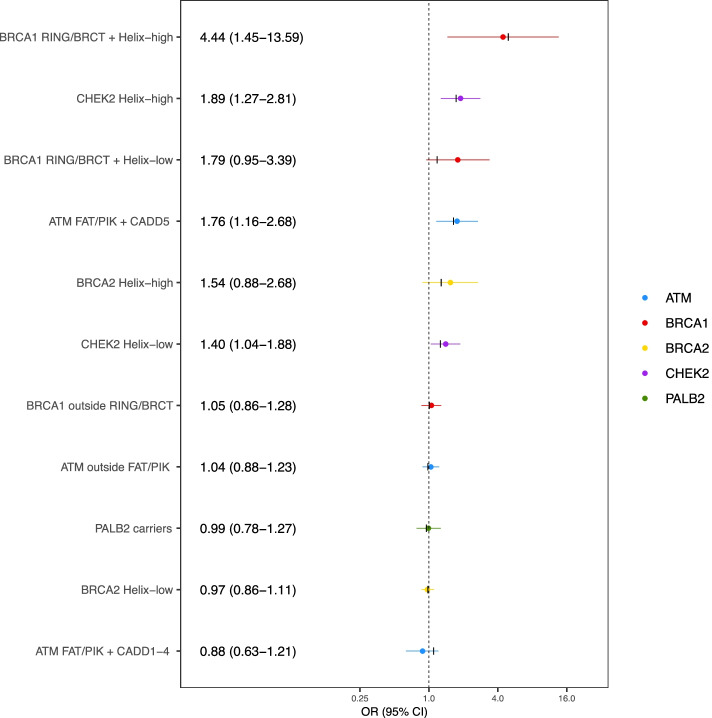
The most predictive in silico algorithms were Helix (BRCA1, BRCA2 and CHEK2) and CADD (ATM). Increased risks appeared restricted to functional protein domains for ATM (FAT and PIK domains) and BRCA1 (RING and BRCT domains). For ATM, BRCA1, and BRCA2, data were compatible with small subsets (approximately 7%, 2%, and 0.6%, respectively) of rare missense variants giving similar risk to those of protein truncating variants in the same gene. For CHEK2, data were more consistent with a large fraction (approximately 60%) of rare missense variants giving a lower risk (OR 1.75, 95% CI (1.47-2.08)) than CHEK2 protein truncating variants. There was little evidence for an association with risk for missense variants in PALB2. The best fitting models were well calibrated in the validation set.
These results will inform risk prediction models and the selection of candidate variants for functional assays and could contribute to the clinical reporting of gene panel testing for breast cancer susceptibility.
ATM、BRCA1、BRCA2、CHEK2 和 PALB2 中的蛋白截断变异与乳腺癌风险增加相关,但这些基因中错义变异的相关风险尚不确定。
我们分析了参与乳腺癌协会联盟 BRIDGES 项目的研究中 59639 例乳腺癌病例和 53165 例对照的数据。我们对 80%的训练集(training set)和 20%的验证集(validation set)进行抽样,以分析 ATM(1146 个训练变体)、BRCA1(644 个)、BRCA2(1425 个)、CHEK2(325 个)和 PALB2(472 个)中的罕见错义变异。我们使用逻辑回归模型和混合模型(假设其中一部分变体与风险相关),根据五种致病变异预测算法、功能蛋白域和频率评估乳腺癌风险。
最具预测性的计算算法是 Helix(BRCA1、BRCA2 和 CHEK2)和 CADD(ATM)。风险增加似乎仅限于 ATM(FAT 和 PIK 结构域)和 BRCA1(RING 和 BRCT 结构域)的功能蛋白结构域。对于 ATM、BRCA1 和 BRCA2,数据与同一基因中蛋白截断变异体具有相似风险的罕见错义变异体的小亚群(分别约为 7%、2%和 0.6%)兼容。对于 CHEK2,数据更符合给予较低风险(OR 1.75,95%CI(1.47-2.08))的大量罕见错义变异体(约 60%),而不是 CHEK2 蛋白截断变异体。PALB2 错义变异体与风险无关联。验证集中最佳拟合模型校准良好。
这些结果将为风险预测模型提供信息,并为功能检测候选变异体的选择提供信息,可能有助于对乳腺癌易感性基因面板检测的临床报告。