From the University of Kansas Medical Center (M.M.D., R.J.B.), Kansas City; University of Missouri (R.J.B.), Columbia; University of Florida (B.B.), Gainesville; LDRTC (O.G.-A.), Fairfax, VA; Duke University Medical Center (P.S.K., L.D.M.P.), Durham, NC; Barrow Neurological Institute (S.L.), Phoenix, AZ; Centre de Référence des Maladies Neuromusculaires Nord/Est/Ile de France Service de Neurologie (P.L.), Hôpital Raymond-Poincaré, Garches, AP-HP and INSERM U1179, Université Versailles Saint-Quentin-en-Yvelines, Montigny-le-Bretonneux; SphinCS GmbH (K.E.M.), Institute of Clinical Science for LSD, Hochheim, Germany; Cincinnati Children's Hospital Medical Center and University of Cincinnati College of Medicine (L.D.M.P.), OH; Neuromuscular Diseases Centre (S.S.), Department of Clinical Neurosciences, University Hospital of Nice (CHU), France; Newcastle University John Walton Muscular Dystrophy Research Centre (V.S.), Newcastle Hospitals NHS Foundation Trust, United Kingdom; University of Texas Southwestern Medical Center (J.T.), Dallas; Department of Neurosciences (P.V.D.), KU Leuven (Catholic University of Leuven), VIB-Center for Brain & Disease Research, and Department of Neurology, University Hospitals Leuven, Belgium; Erasmus MC University Medical Center (A.T.v.d.P.), Pompe Center & Center for Lysosomal and Metabolic Diseases, Rotterdam, the Netherlands; Copenhagen Neuromuscular Center (J.V.), Rigshospitalet, University of Copenhagen, Denmark; Department of Neurology (P.Y.), Medical Park Bad Feilnbach, Germany; Sanofi (K.A.H., P.M.), Chilly-Mazarin, France; Sanofi (M.F., T.Z.), Cambridge, MA; Elevate Medical Affairs (J.M.G.), Horsham, United Kingdom; Sanofi (O.V.), Montpellier, France; and Friedrich-Baur-Institut (B.S.), Department of Neurology Klinikum München, Germany.
Neurology. 2022 Aug 1;99(5):e536-e548. doi: 10.1212/WNL.0000000000200746.
Pompe disease is a rare, progressive neuromuscular disorder caused by deficiency of lysosomal acid α-glucosidase (GAA) and subsequent glycogen accumulation. Avalglucosidase alfa, a recombinant human GAA enzyme replacement therapy designed for increased cellular uptake and glycogen clearance, has been studied for long-term efficacy and safety in patients with late-onset Pompe disease (LOPD). Here, we report up to 6.5 years' experience with avalglucosidase alfa during the NEO1 and NEO-EXT studies.
NEO1 participants with LOPD, either treatment naive (Naive Group) or receiving alglucosidase alfa for ≥9 months (Switch Group), received avalglucosidase alfa (5, 10, or 20 mg/kg every other week [qow]) for 6 months before entering NEO-EXT and continued their NEO1 dose until all proceeded with 20 mg/kg qow. Safety and efficacy, a prespecified exploratory secondary outcome, were assessed; slopes of change for efficacy outcomes were calculated from a repeated mixed-measures model.
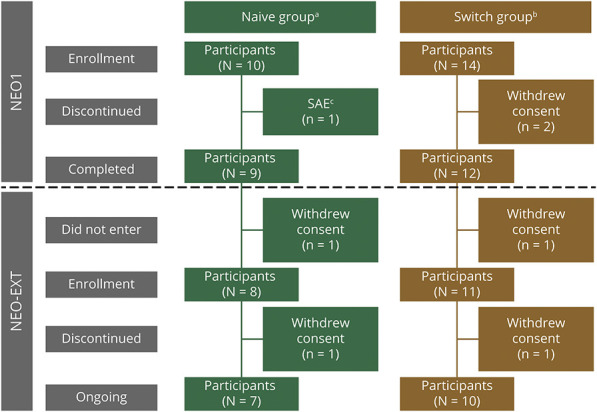
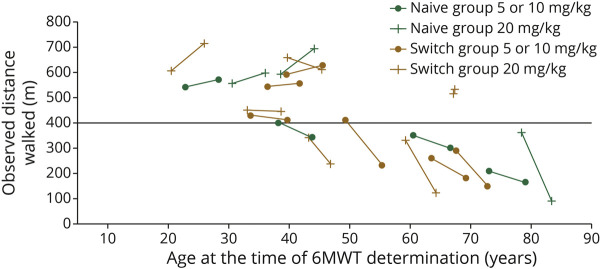
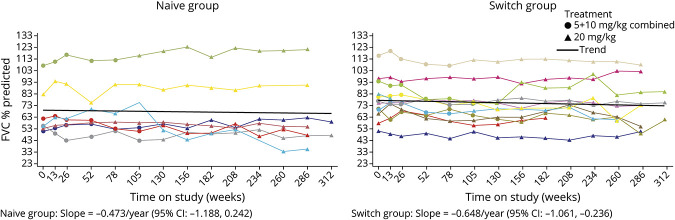
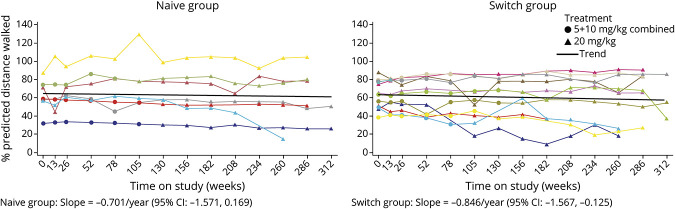
Twenty-four participants enrolled in NEO1 (Naive Group, n = 10; Switch Group, n = 14); 21 completed and 19 entered NEO-EXT; in February 2020, 17 participants remained in NEO-EXT, with data up to 6.5 years. Avalglucosidase alfa was generally well tolerated during NEO-EXT, with a safety profile consistent with that in NEO1. No deaths or treatment-related life-threatening serious adverse events occurred. Eighteen participants developed antidrug antibodies without apparent effect on clinical outcomes. No participants who were tested developed immunoglobulin E antibodies. Upright forced vital capacity %predicted remained stable in most participants, with slope estimates (95% CIs) of -0.473 per year (-1.188 to 0.242) and -0.648 per year (-1.061 to -0.236) in the Naive and Switch Groups, respectively. Six-minute walk test (6MWT) %predicted was also stable for most participants, with slope estimates of -0.701 per year (-1.571 to 0.169) and -0.846 per year (-1.567 to -0.125) for the Naive and Switch Groups, respectively. Improvements in 6MWT distance were observed in most participants aged <45 years at NEO1 enrollment in both the Naive and Switch Groups.
Avalglucosidase alfa was generally well tolerated for up to 6.5 years in adult participants with LOPD either naive to alglucosidase alfa or who had previously received alglucosidase alfa for ≥9 months.
This study provides Class IV evidence of long-term tolerability and sustained efficacy of avalglucosidase alfa in patients with LOPD after up to 6.5 years.
NCT01898364 (NEO1 first posted: July 12, 2013; clinicaltrials.gov/ct2/show/NCT01898364); NCT02032524 (NEO-EXT first posted: January 10, 2014; clinicaltrials.gov/ct2/show/NCT02032524). First participant enrollment: NEO1-August 19, 2013; NEO-EXT-February 27, 2014.
庞贝病是一种罕见的进行性神经肌肉疾病,由溶酶体酸性α-葡萄糖苷酶(GAA)缺乏引起,随后发生糖原积累。阿伐糖苷酶 α 是一种重组人 GAA 酶替代疗法,旨在增加细胞摄取和糖原清除,已在晚发性庞贝病(LOPD)患者中进行了长期疗效和安全性研究。在此,我们报告了在 NEO1 和 NEO-EXT 研究中使用阿伐糖苷酶 α 的长达 6.5 年的经验。
NEO1 中患有 LOPD 的参与者,无论是治疗初治(初治组)还是接受阿伐糖苷酶 α 治疗≥9 个月(转换组),在进入 NEO-EXT 前接受 6 个月的阿伐糖苷酶 α(每隔一周 5、10 或 20 mg/kg)治疗,然后继续使用 NEO1 剂量,直到所有参与者都使用 20 mg/kg qow。评估了安全性和疗效,这是预先指定的探索性次要终点;从重复混合效应模型计算了疗效结果的变化斜率。
24 名参与者参加了 NEO1(初治组,n=10;转换组,n=14);21 名完成并 19 名进入 NEO-EXT;2020 年 2 月,17 名参与者仍在 NEO-EXT 中,数据最长达 6.5 年。在 NEO-EXT 期间,阿伐糖苷酶 α 通常耐受性良好,安全性与 NEO1 一致。没有死亡或与治疗相关的危及生命的严重不良事件发生。18 名参与者产生了抗药物抗体,但对临床结果没有明显影响。没有参与者测试出免疫球蛋白 E 抗体。大多数参与者的直立用力肺活量%预测值保持稳定,初治组和转换组的斜率估计值(95%CI)分别为每年-0.473(-1.188 至 0.242)和每年-0.648(-1.061 至-0.236)。大多数参与者的 6 分钟步行试验(6MWT)%预测值也保持稳定,初治组和转换组的斜率估计值分别为每年-0.701(-1.571 至 0.169)和每年-0.846(-1.567 至-0.125)。在 NEO1 入组时年龄<45 岁的大多数参与者中,6MWT 距离有所改善,初治组和转换组均如此。
在患有 LOPD 的成年参与者中,阿伐糖苷酶 α 的耐受性通常良好,最长可达 6.5 年,这些参与者要么对阿伐糖苷酶 α 初治,要么此前接受过阿伐糖苷酶 α 治疗≥9 个月。
这项研究提供了 IV 级证据,表明在长达 6.5 年的时间里,阿伐糖苷酶 α 在患有 LOPD 的患者中具有长期耐受性和持续疗效。
NCT01898364(NEO1 首次发布:2013 年 7 月 12 日;clinicaltrials.gov/ct2/show/NCT01898364);NCT02032524(NEO-EXT 首次发布:2014 年 1 月 10 日;clinicaltrials.gov/ct2/show/NCT02032524)。首例参与者入组:NEO1-2013 年 8 月 19 日;NEO-EXT-2014 年 2 月 27 日。