Chimusa Emile R, Defo Joel
Division of Human Genetics, Department of Pathology, Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Cape Town, South Africa.
Front Genet. 2022 May 18;13:838518. doi: 10.3389/fgene.2022.838518. eCollection 2022.
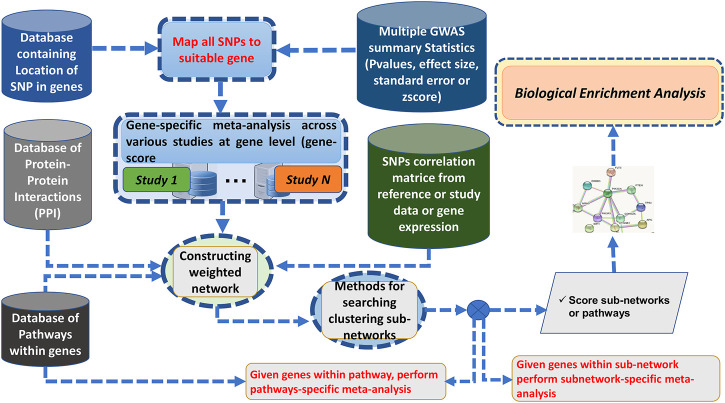
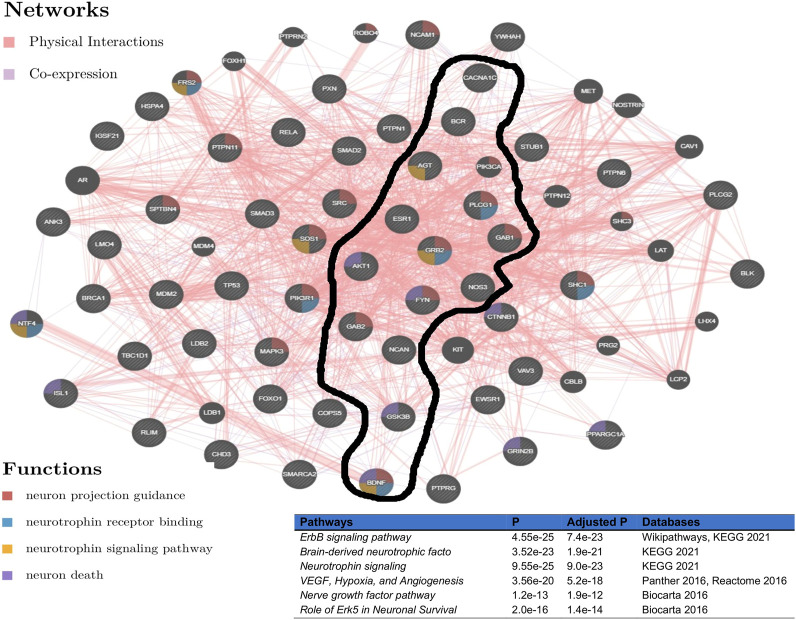
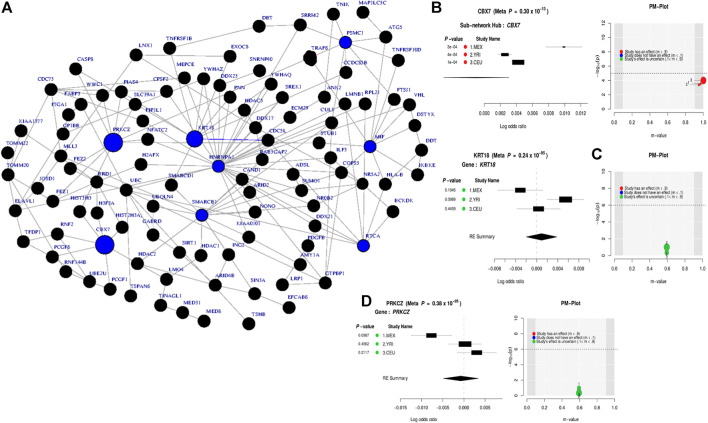
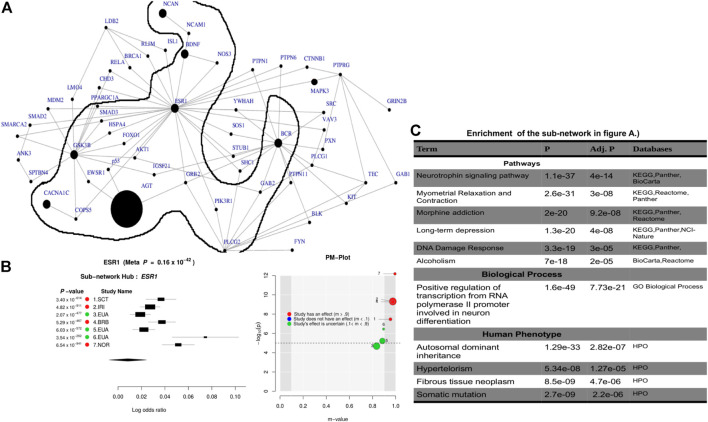
Over the past decades, advanced high-throughput technologies have continuously contributed to genome-wide association studies (GWASs). GWAS meta-analysis has been increasingly adopted, has cross-ancestry replicability, and has power to illuminate the genetic architecture of complex traits, informing about the reliability of estimation effects and their variability across human ancestries. However, detecting genetic variants that have low disease risk still poses a challenge. Designing a meta-analysis approach that combines the effect of various SNPs within genes or genes within pathways from multiple independent population GWASs may be helpful in identifying associations with small effect sizes and increasing the association power. Here, we proposed ancMETA, a Bayesian graph-based framework, to perform the gene/pathway-specific meta-analysis by combining the effect size of multiple SNPs within genes, and genes within subnetwork/pathways across multiple independent population GWASs to deconvolute the interactions between genes underlying the pathogenesis of complex diseases across human populations. We assessed the proposed framework on simulated datasets, and the results show that the proposed model holds promise for increasing statistical power for meta-analysis of genetic variants underlying the pathogenesis of complex diseases. To illustrate the proposed meta-analysis framework, we leverage seven different European bipolar disorder (BD) cohorts, and we identify variants in the angiotensinogen () gene to be significantly associated with BD across all 7 studies. We detect a commonly significant BD-specific subnetwork with the gene as the main hub of a subnetwork, associated with neurotrophin signaling (p = 4 ) and myometrial relaxation and contraction (p = 3 ) pathways. ancMETA provides a new contribution to post-GWAS methodologies and holds promise for comprehensively examining interactions between genes underlying the pathogenesis of genetic diseases and also underlying ethnic differences.
在过去几十年中,先进的高通量技术不断推动全基因组关联研究(GWAS)的发展。GWAS荟萃分析越来越多地被采用,具有跨祖先的可重复性,并且有能力阐明复杂性状的遗传结构,为估计效应的可靠性及其在人类不同祖先群体中的变异性提供信息。然而,检测低疾病风险的遗传变异仍然是一个挑战。设计一种荟萃分析方法,将来自多个独立人群GWAS的基因内各种单核苷酸多态性(SNP)或通路内基因的效应结合起来,可能有助于识别小效应量的关联并提高关联效能。在此,我们提出了ancMETA,一个基于贝叶斯图的框架,通过结合基因内多个SNP以及多个独立人群GWAS的子网/通路内基因的效应大小,来进行基因/通路特异性的荟萃分析,以解析人类群体中复杂疾病发病机制背后基因之间的相互作用。我们在模拟数据集上评估了所提出的框架,结果表明该模型有望提高对复杂疾病发病机制潜在遗传变异进行荟萃分析的统计效能。为了说明所提出的荟萃分析框架,我们利用了七个不同欧洲双相情感障碍(BD)队列,并在所有七项研究中确定血管紧张素原()基因中的变异与BD显著相关。我们检测到一个普遍显著的BD特异性子网,该子网以基因作为主要枢纽,与神经营养因子信号传导(p = 4)和子宫肌层舒张与收缩(p = 3)通路相关。ancMETA为GWAS后方法学做出了新贡献,有望全面研究遗传疾病发病机制背后基因之间的相互作用以及潜在的种族差异。