Aboelnga Mohamed M
Chemistry Department, Faculty of Science, Damietta University New Damietta 34517 Egypt
RSC Adv. 2022 May 23;12(24):15543-15554. doi: 10.1039/d2ra01073a. eCollection 2022 May 17.
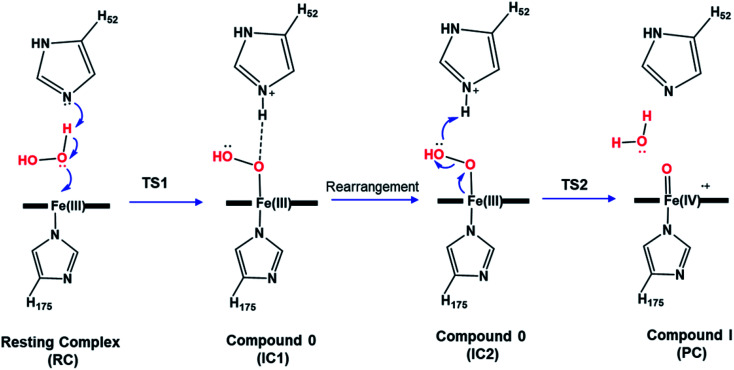
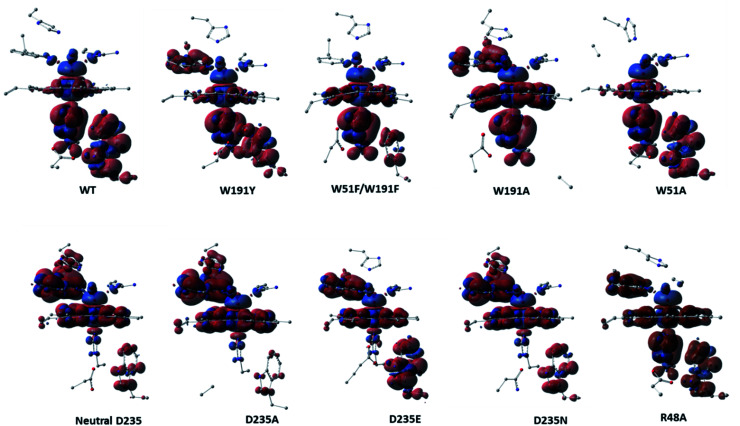
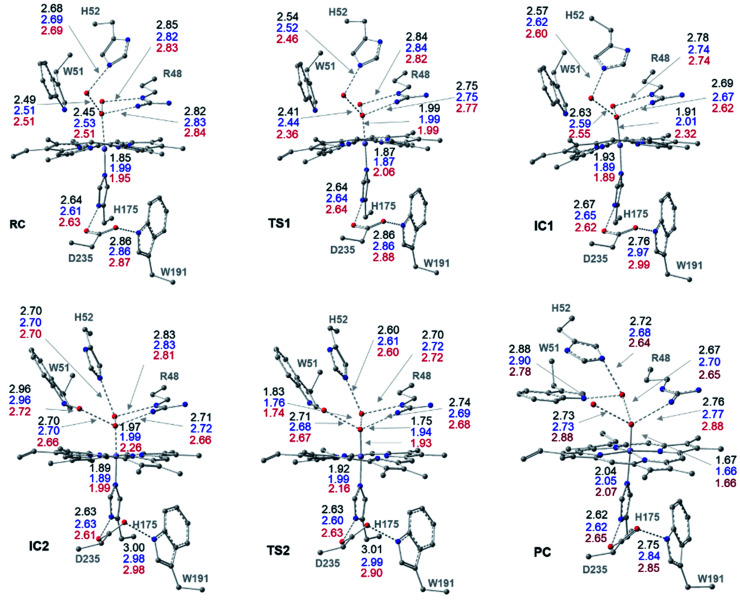
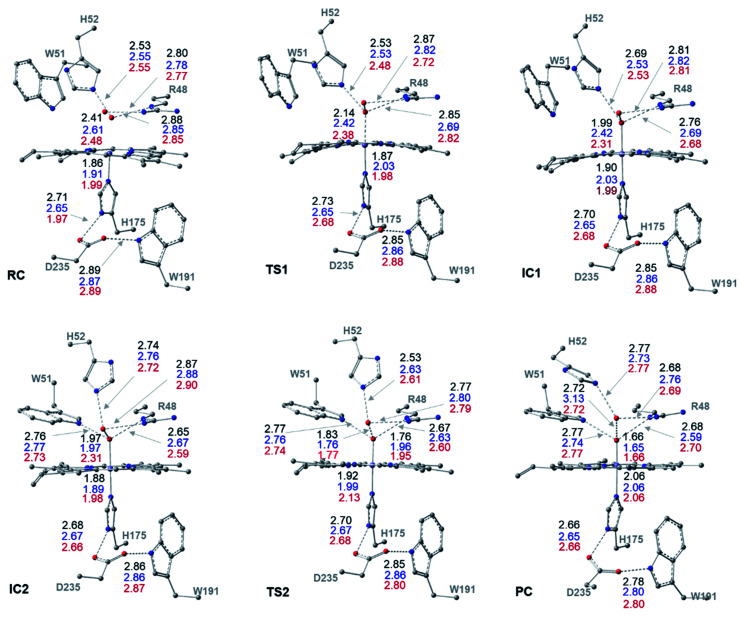
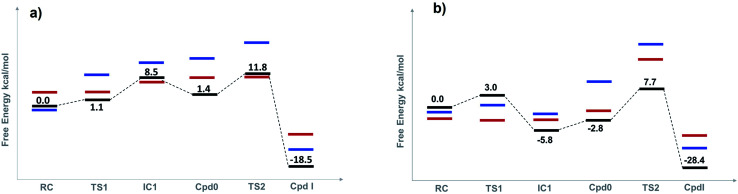
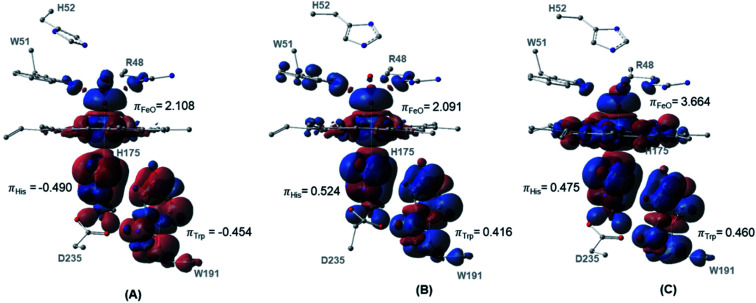
Peroxidases are heme containing enzymes that catalyze peroxide-dependant oxidation of a variety of substrates through forming key ferryl intermediates, compounds I and II. Cytochrome peroxidase (Ccp1) has served for decades as a chemical model toward understanding the chemical biology of this heme family of enzymes. It is known to feature a distinctive electronic behaviour for its compound I despite significant structural similarity to other peroxidases. A water-assisted mechanism has been proposed over a dry one for the formation of compound I in similar peroxidases. To better identify the viability of these mechanisms, we employed quantum chemistry calculations for the heme pocket of Ccp1 in three different spin states. We provided comparative energetic and structural results for the six possible pathways that suggest the preference of the dry mechanism energetically and structurally. The doublet state is found to be the most preferable spin state for the mechanism to proceed and for the formation of the Cpd I ferryl-intermediate irrespective of the considered dielectric constant used to represent the solvent environment. The nature of the spin state has negligible effects on the calculated structures but great impact on the energetics. Our analysis was also expanded to explain the major contribution of key residues to the peroxidase activity of Ccp1 through exploring the mechanism at various generated Ccp1 variants. Overall, we provide valuable findings toward solving the current ambiguity of the exact mechanism in Ccp1, which could be applied to peroxidases with similar heme pockets.
过氧化物酶是含血红素的酶,通过形成关键的高铁中间体、化合物I和化合物II,催化多种底物的过氧化物依赖性氧化。几十年来,细胞色素过氧化物酶(Ccp1)一直作为一种化学模型,用于理解这类血红素酶家族的化学生物学。尽管与其他过氧化物酶在结构上有显著相似性,但已知其化合物I具有独特的电子行为。对于类似过氧化物酶中化合物I的形成,有人提出了一种水辅助机制,而非无水机制。为了更好地确定这些机制的可行性,我们对处于三种不同自旋态的Ccp1血红素口袋进行了量子化学计算。我们给出了六种可能途径的能量和结构比较结果,表明在能量和结构上无水机制更具优势。无论用于表示溶剂环境的介电常数如何,发现双重态是该机制进行以及形成Cpd I高铁中间体的最优选自旋态。自旋态的性质对计算出的结构影响可忽略不计,但对能量学有很大影响。我们的分析还通过探索各种生成的Ccp1变体的机制,扩展到解释关键残基对Ccp1过氧化物酶活性的主要贡献。总体而言,我们为解决目前Ccp1确切机制的模糊性提供了有价值的发现,这些发现可应用于具有类似血红素口袋的过氧化物酶。