Corcoran Daniel, Maltbie Nick, Sudalairaj Shivchander, Baker Frazier N, Hirschfeld Joseph, Porollo Aleksey
Department of Electrical Engineering and Computing Systems, University of Cincinnati, Cincinnati, OH, USA.
Advanced Concepts Laboratory, Georgia Tech Research Institute, Fairborn, OH, USA.
Front Bioinform. 2021;1. doi: 10.3389/fbinf.2021.653681. Epub 2021 Jun 24.
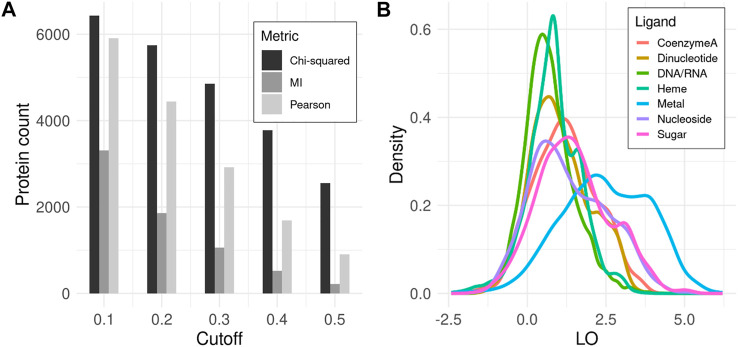
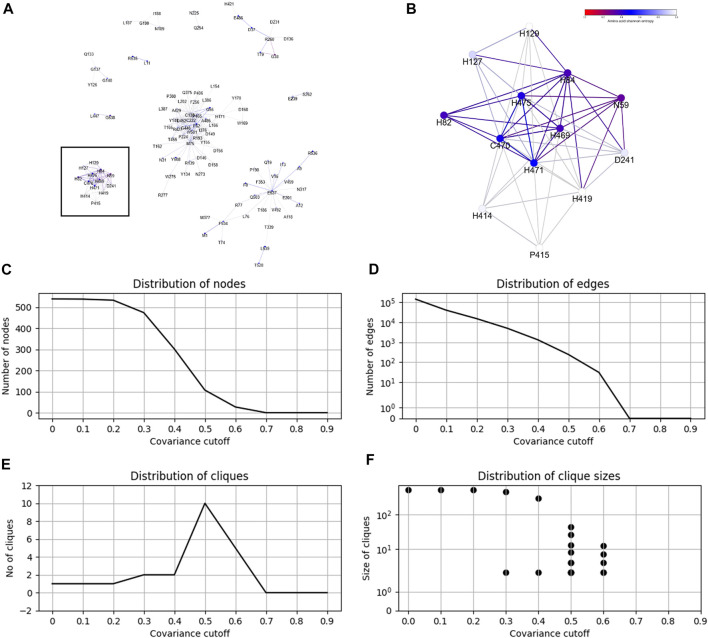
Proteins by and large carry out their molecular functions in a folded state when residues, distant in sequence, assemble together in 3D space to bind a ligand, catalyze a reaction, form a channel, or exert another concerted macromolecular interaction. It has been long recognized that covariance of amino acids between distant positions within a protein sequence allows for the inference of long range contacts to facilitate 3D structure modeling. In this work, we investigated whether covariance analysis may reveal residues involved in the same molecular function. Building upon our previous work, CoeViz, we have conducted a large scale covariance analysis among 7595 non-redundant proteins with resolved 3D structures to assess (1) whether the residues with the same function coevolve, (2) which covariance metric captures such couplings better, and (3) how different molecular functions compare in this context. We found that the chi-squared metric is the most informative for the identification of coevolving functional sites, followed by the Pearson correlation-based, whereas mutual information is the least informative. Of the seven categories of the most common natural ligands, including coenzyme A, dinucleotide, DNA/RNA, heme, metal, nucleoside, and sugar, the trace metal binding residues display the most prominent coupling, followed by the sugar binding sites. We also developed a web-based tool, CoeViz 2, that enables the interactive visualization of covarying residues as cliques from a larger protein graph. CoeViz 2 is publicly available at https://research.cchmc.org/CoevLab/.
一般来说,蛋白质在折叠状态下执行其分子功能,此时序列上相隔较远的残基在三维空间中聚集在一起,以结合配体、催化反应、形成通道或发挥其他协同的大分子相互作用。长期以来,人们已经认识到蛋白质序列中远距离位置之间氨基酸的协方差有助于推断长程接触,从而促进三维结构建模。在这项工作中,我们研究了协方差分析是否可以揭示参与相同分子功能的残基。基于我们之前的工作CoeViz,我们对7595个具有解析三维结构的非冗余蛋白质进行了大规模协方差分析,以评估:(1)具有相同功能的残基是否共同进化;(2)哪种协方差度量能更好地捕捉这种耦合;(3)在这种情况下不同分子功能如何比较。我们发现,卡方度量对于识别共同进化的功能位点最具信息性,其次是基于皮尔逊相关的度量,而互信息的信息性最差。在七种最常见的天然配体类别中,包括辅酶A、二核苷酸、DNA/RNA、血红素、金属、核苷和糖,微量金属结合残基表现出最显著的耦合,其次是糖结合位点。我们还开发了一个基于网络的工具CoeViz 2,它能够将共同变化的残基作为来自更大蛋白质图的团进行交互式可视化。CoeViz 2可在https://research.cchmc.org/CoevLab/上公开获取。