Firoz Ahmad, Malik Adeel, Afzal Obaid, Jha Vivekanand
Bioinformation. 2010 Jul 6;5(2):55-7. doi: 10.6026/97320630005055.
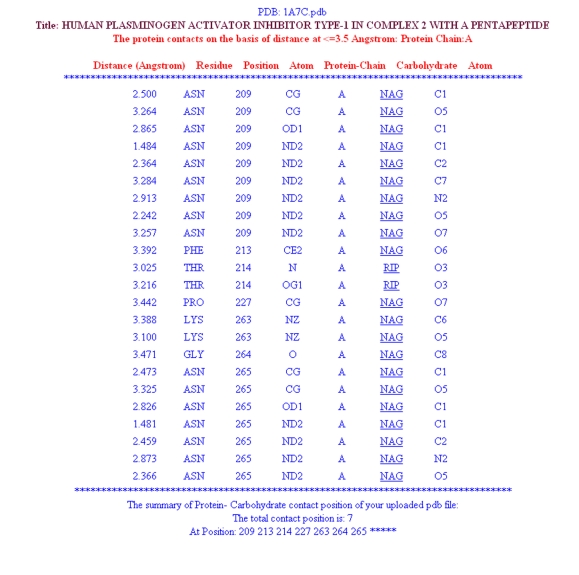
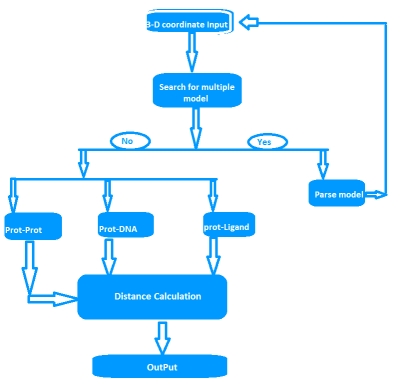
To investigate the functional sites on a protein and the prediction of binding sites (residues)in proteins, it is often required to identify the binding site residues at different distance threshold from protein three dimensional (3D)structures. For the study of a particular protein chain and its interaction with the ligand in complex form, researchers have to parse the output of different available tools or databases for finding binding-site residues. Here we have developed a tool for calculating amino acid contact distances in proteins at different distance threshold from the 3D-structure of the protein. For an input of protein 3D-structure, ContPro can quickly find all binding-site residues in the protein by calculating distances and also allows researchers to select the different distance threshold, protein chain and ligand of interest. Additionally, it can also parse the protein model (in case of multi model protein coordinate file)and the sequence of selected protein chain in Fasta format from the input 3D-structure. The developed tool will be useful for the identification and analysis of binding sites of proteins from 3D-structure at different distance thresholds.
IT CAN BE ACCESSED AT: http://procarb.org/contpro/
为了研究蛋白质上的功能位点以及预测蛋白质中的结合位点(残基),通常需要从蛋白质三维(3D)结构中识别不同距离阈值下的结合位点残基。对于特定蛋白质链及其与配体以复合物形式相互作用的研究,研究人员必须解析不同可用工具或数据库的输出以找到结合位点残基。在此,我们开发了一种工具,用于根据蛋白质的3D结构计算不同距离阈值下蛋白质中氨基酸的接触距离。对于蛋白质3D结构的输入,ContPro可以通过计算距离快速找到蛋白质中的所有结合位点残基,还允许研究人员选择感兴趣的不同距离阈值、蛋白质链和配体。此外,它还可以从输入的3D结构中解析蛋白质模型(如果是多模型蛋白质坐标文件)以及以Fasta格式表示的所选蛋白质链的序列。所开发的工具将有助于在不同距离阈值下从3D结构识别和分析蛋白质的结合位点。
可通过以下网址访问:http://procarb.org/contpro/