Medical Genetics Unit and Thalassemia Center, San Luigi University Hospital, University of Torino, Orbassano, TO, Italy.
Institute of Human Genetics, Center for Molecular Medicine Cologne, and Center for Rare and Hereditary Kidney Disease, University Hospital of Cologne, CologneCologne, Germany.
Pediatr Nephrol. 2023 Mar;38(3):625-634. doi: 10.1007/s00467-022-05613-2. Epub 2022 Jun 13.
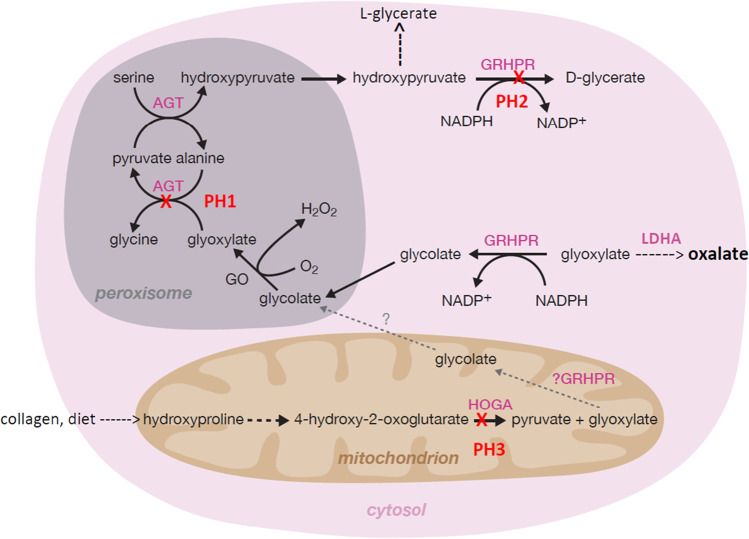
Accurate diagnosis of primary hyperoxaluria (PH) has important therapeutic consequences. Since biochemical assessment can be unreliable, genetic testing is a crucial diagnostic tool for patients with PH to define the disease type. Patients with PH type 1 (PH1) have a worse prognosis than those with other PH types, despite the same extent of oxalate excretion. The relation between genotype and clinical phenotype in PH1 is extremely heterogeneous with respect to age of first symptoms and development of kidney failure. Some mutations are significantly linked to pyridoxine-sensitivity in PH1, such as homozygosity for p.G170R and p.F152I combined with a common polymorphism. Although patients with these mutations display on average better outcomes, they may also present with CKD stage 5 in infancy. In vitro studies suggest pyridoxine-sensitivity for some other mutations, but confirmatory clinical data are lacking (p.G47R, p.G161R, p.I56N/major allele) or scarce (p.I244T). These studies also suggest that other vitamin B6 derivatives than pyridoxine may be more effective and should be a focus for clinical testing. PH patients displaying the same mutation, even within one family, may have completely different clinical outcomes. This discordance may be caused by environmental or genetic factors that are unrelated to the effect of the causative mutation(s). No relation between genotype and clinical or biochemical phenotypes have been found so far in PH types 2 and 3. This manuscript reviews the current knowledge on the genetic background of the three types of primary hyperoxaluria and its impact on clinical management, including prenatal diagnosis.
准确诊断原发性高草酸尿症 (PH) 具有重要的治疗意义。由于生化评估可能不可靠,因此基因检测是 PH 患者的重要诊断工具,可确定疾病类型。PH 1 型 (PH1) 患者的预后比其他 PH 类型差,尽管草酸排泄量相同。PH1 中基因型与临床表型之间的关系在首发症状和肾衰竭的发展方面存在极大的异质性。某些突变与 PH1 中的吡哆醇敏感性显著相关,例如 p.G170R 和 p.F152I 的纯合性与常见的多态性相结合。尽管这些突变患者的平均预后较好,但他们也可能在婴儿期出现 CKD 5 期。体外研究表明,一些其他突变也与吡哆醇敏感性相关,但缺乏确认性临床数据(p.G47R、p.G161R、p.I56N/主要等位基因)或数据稀少(p.I244T)。这些研究还表明,除了吡哆醇之外,其他维生素 B6 衍生物可能更有效,应成为临床检测的重点。即使在一个家庭中,具有相同突变的 PH 患者也可能具有完全不同的临床结局。这种不一致可能是由与致病突变无关的环境或遗传因素引起的。迄今为止,在 PH 2 型和 3 型中尚未发现基因型与临床或生化表型之间的关系。本文综述了三种原发性高草酸尿症的遗传背景及其对临床管理的影响,包括产前诊断。