Department of Pharmacology, University of Oxford, Oxford, UK.
Structural Bioinformatics and Computational Biochemistry, Department of Biochemistry, University of Oxford, Oxford, UK.
Br J Pharmacol. 2022 Nov;179(21):4941-4957. doi: 10.1111/bph.15893. Epub 2022 Aug 2.
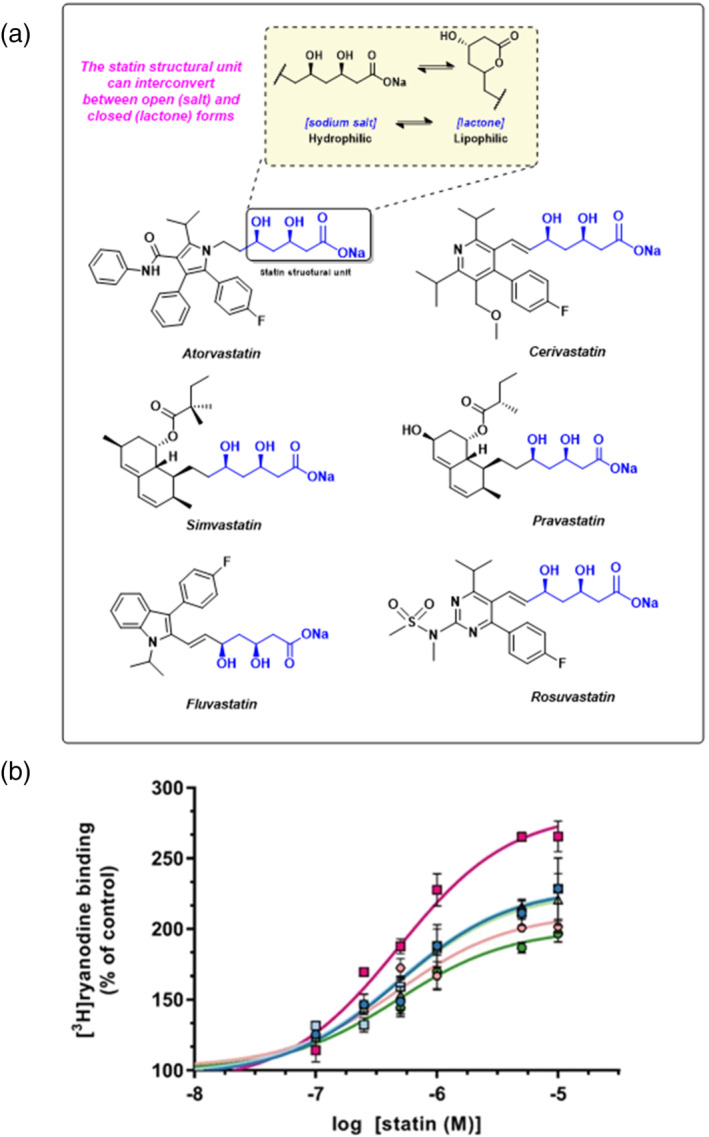
Statins, inhibitors of HMG-CoA reductase, are mainstay treatment for hypercholesterolaemia. However, muscle pain and weakness prevent many patients from benefiting from their cardioprotective effects. We previously demonstrated that simvastatin activates skeletal ryanodine receptors (RyR1), an effect that could be important in initiating myopathy. Using a range of structurally diverse statin analogues, we examined structural features associated with RyR1 activation, aiming to identify statins lacking this property.
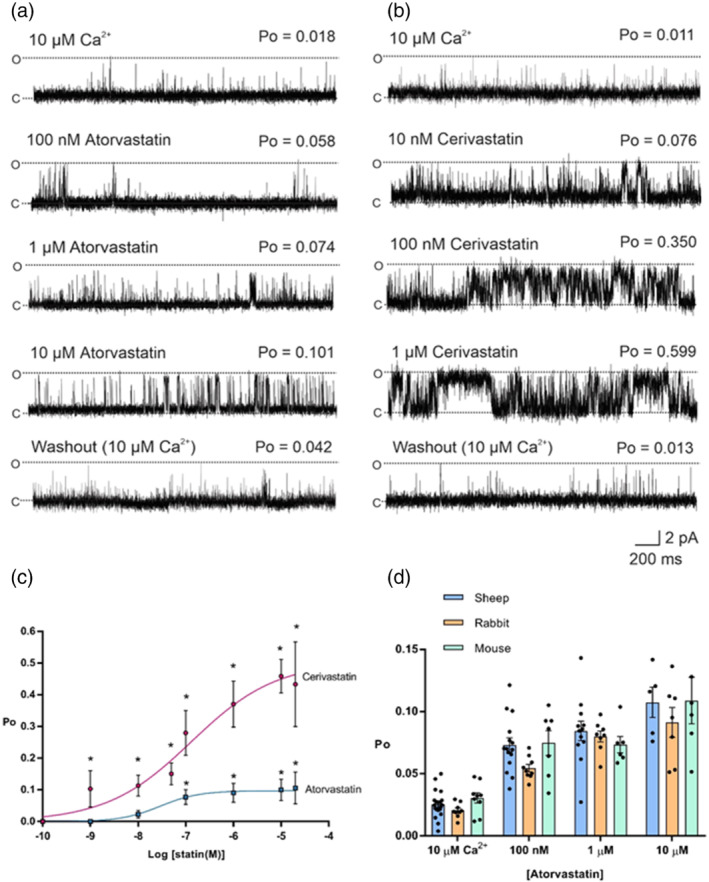
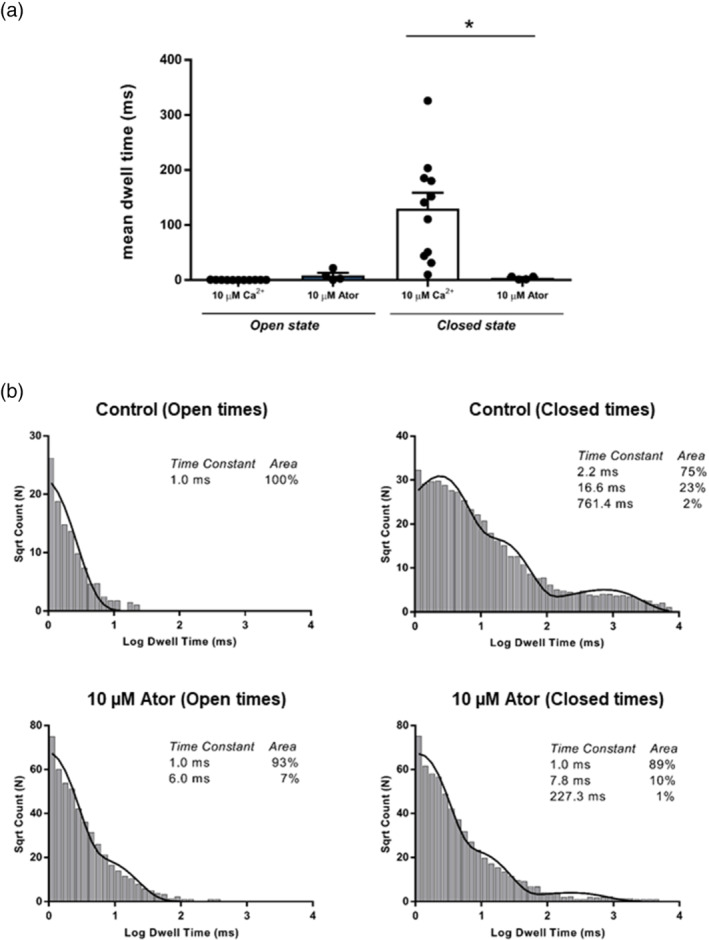
Compounds were screened for RyR1 activity utilising [ H]ryanodine binding. Mechanistic insight into RyR1 activity was studied by incorporating RyR1 channels from sheep, mouse or rabbit skeletal muscle into bilayers.
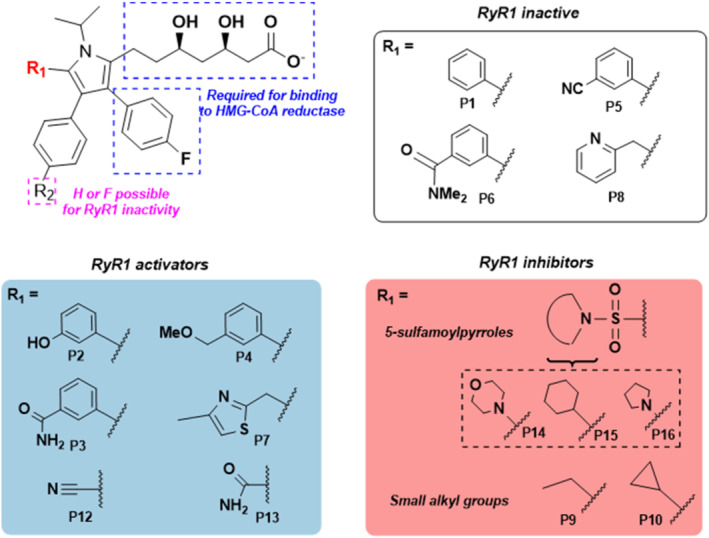
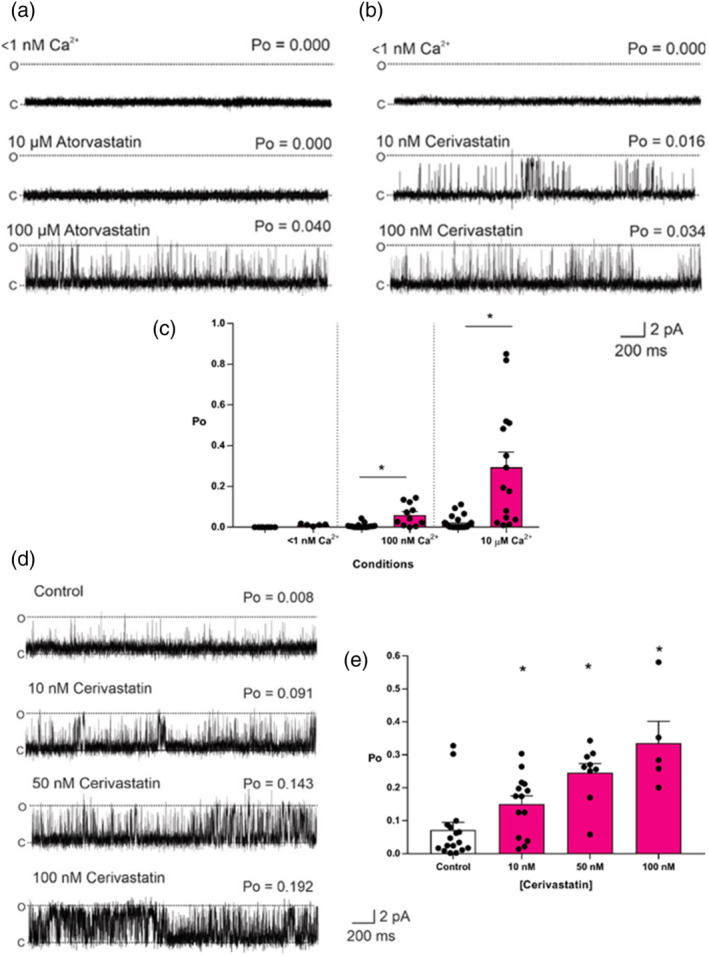
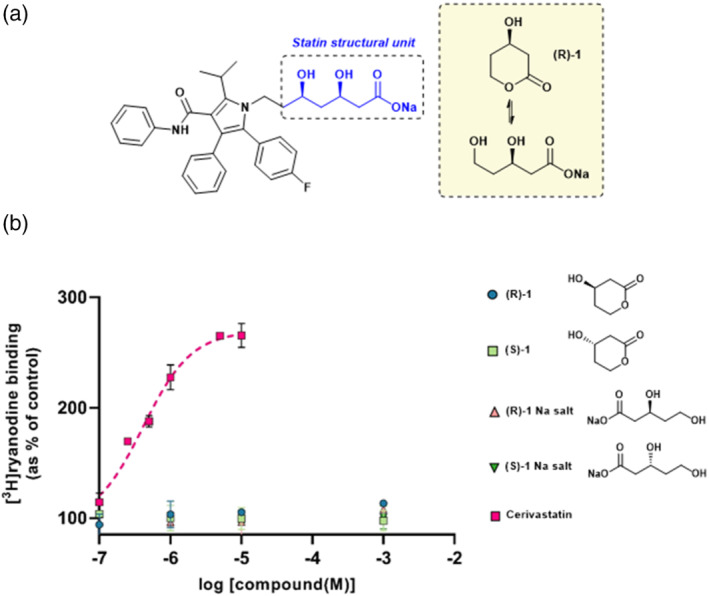
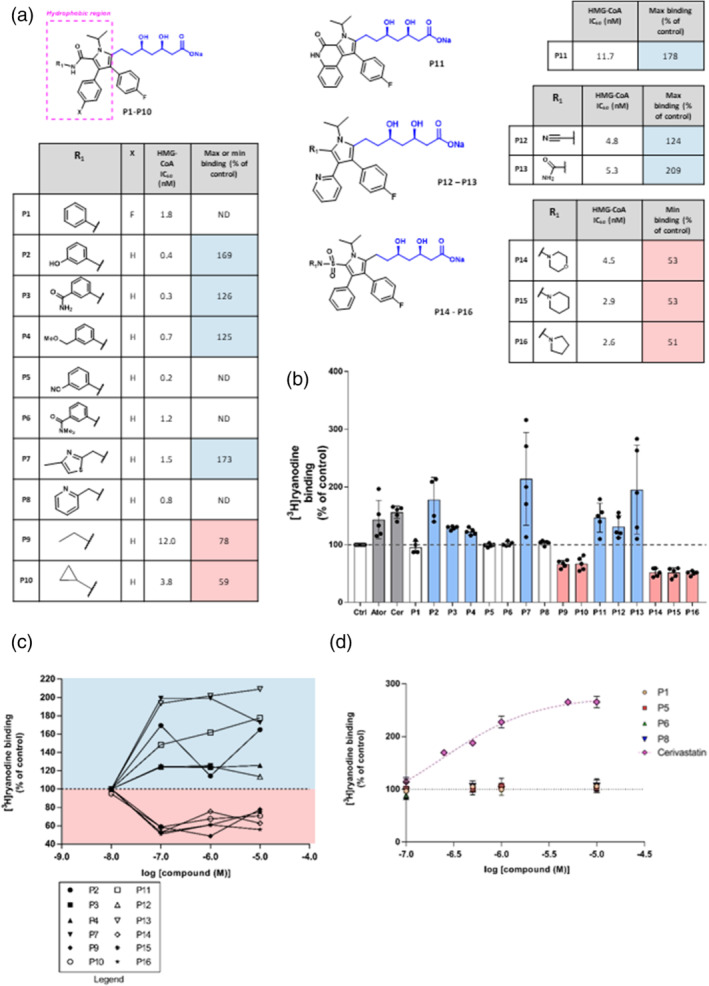
All UK-prescribed statins activated RyR1 at nanomolar concentrations. Cerivastatin, withdrawn from the market due to life-threatening muscle-related side effects, was more effective than currently-prescribed statins and possessed the unique ability to open RyR1 channels independently of cytosolic Ca . We synthesised the one essential structural moiety that all statins must possess for HMG-CoA reductase inhibition, the R-3,5-dihydroxypentanoic acid unit, and it did not activate RyR1. We also identified five analogues retaining potent HMG-CoA reductase inhibition that inhibited RyR1 and four that lacked the ability to modulate RyR1.
That cerivastatin activates RyR1 most strongly supports the hypothesis that RyR1 activation is implicated in statin-induced myopathy. Demonstrating that statin regulation of RyR1 and HMG-CoA reductase are separable effects will allow the role of RyR1 in statin-induced myopathy to be further elucidated by the tool compounds we have identified, allowing development of effective cardioprotective statins with improved patient tolerance.
他汀类药物是 HMG-CoA 还原酶的抑制剂,是治疗高胆固醇血症的主要药物。然而,肌肉疼痛和无力使得许多患者无法从中受益于其心脏保护作用。我们之前的研究表明,辛伐他汀激活了骨骼肌ryanodine 受体(RyR1),这种作用可能在引发肌病方面很重要。本研究使用一系列结构多样的他汀类药物类似物,研究了与 RyR1 激活相关的结构特征,旨在确定缺乏这种特性的他汀类药物。
利用 [ H]ryanodine 结合来筛选化合物对 RyR1 的活性。通过将来自绵羊、小鼠或兔骨骼肌的 RyR1 通道掺入双层膜中来研究 RyR1 活性的机制。
所有英国处方的他汀类药物都以纳摩尔浓度激活 RyR1。因危及生命的肌肉相关副作用而被撤出市场的西立伐他汀比目前处方的他汀类药物更有效,并且具有独特的能力,可以在不依赖细胞质 Ca 2+的情况下打开 RyR1 通道。我们合成了他汀类药物抑制 HMG-CoA 还原酶所必需的唯一结构部分,即 R-3,5-二羟基戊酸单元,它不能激活 RyR1。我们还确定了五种保留强效 HMG-CoA 还原酶抑制作用的类似物,它们抑制 RyR1,还有四种缺乏调节 RyR1 能力的类似物。
西立伐他汀最强烈地激活 RyR1,这有力地支持了 RyR1 激活与他汀类药物诱导的肌病有关的假说。证明他汀类药物对 RyR1 的调节作用和 HMG-CoA 还原酶抑制作用是可分离的,这将使我们鉴定的工具化合物进一步阐明 RyR1 在他汀类药物诱导的肌病中的作用,并开发出具有更好的患者耐受性的有效的心脏保护他汀类药物。