Department of Neurology and Geriatrics, Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima, Japan.
Department of Neurology, Imakiire General Hospital, Kagoshima, Japan.
Ann Clin Transl Neurol. 2022 Jul;9(7):902-911. doi: 10.1002/acn3.51603. Epub 2022 Jun 22.
Recessive mutations in SLC12A6 have been linked to hereditary motor sensory neuropathy with agenesis of the corpus callosum. Patients with early-onset peripheral neuropathy associated with SLC12A6 heterozygous variants were reported in 2016. Only five families and three variants have been reported to date, and the spectrum is unclear. Here, we aim to describe the clinical and mutation spectra of SLC12A6-related Charcot-Marie-Tooth (CMT) disease in Japanese patients.
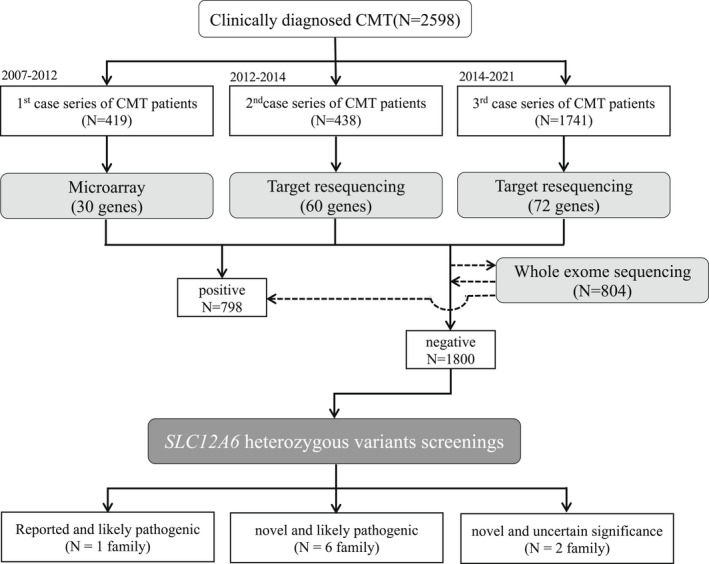
We extracted SLC12A6 variants from our DNA microarray and targeted resequencing data obtained from 2598 patients with clinically suspected CMT who were referred to our genetic laboratory by neurological or neuropediatric departments across Japan. And we summarized the clinical and genetic features of these patients.
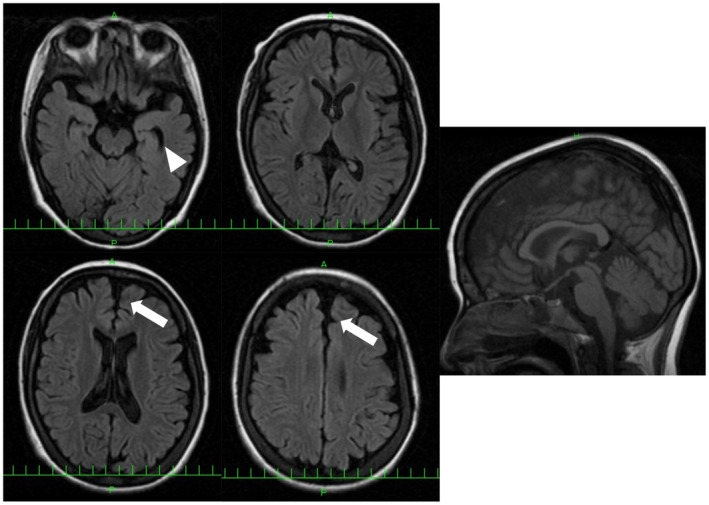
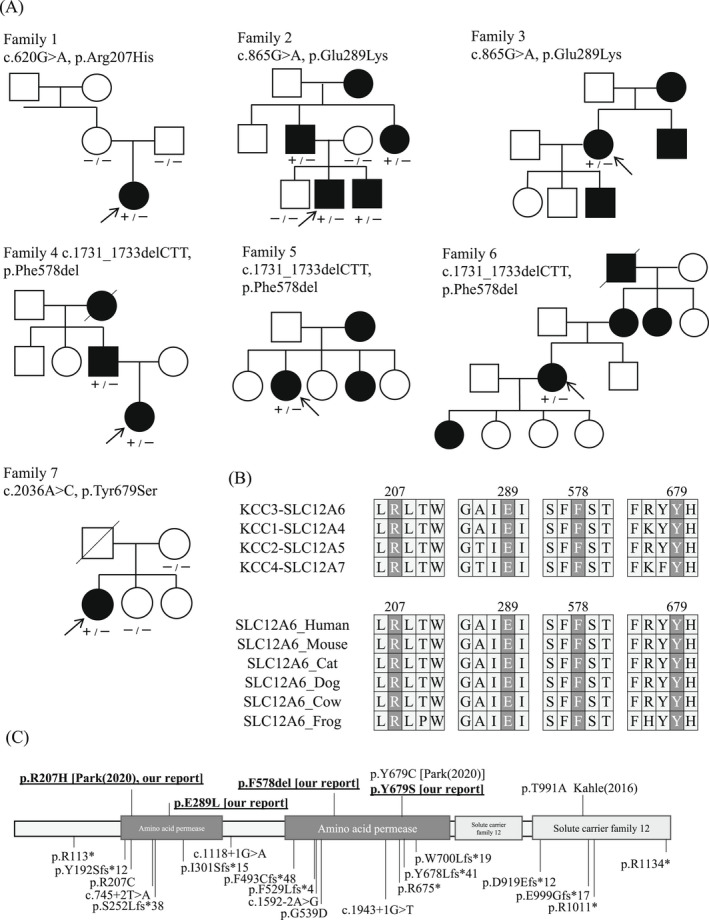
In seven unrelated families, we identified one previously reported and three novel likely pathogenic SLC12A6 heterozygous variants, as well as two variants of uncertain significance. The mean age of onset for these patients was 17.5 ± 16.1 years. Regarding electrophysiology, the median motor nerve conduction velocity was 39.6 ± 9.5 m/sec. For the first time, we observed intellectual disability in three patients. One patient developed epilepsy, and her brain MRI revealed frontal and temporal lobe atrophy without changes in white matter and corpus callosum.
Screening for the SLC12A6 gene should be considered in patients with CMT, particularly those with central nervous system lesions, such as cognitive impairment and epilepsy, regardless of the CMT subtype.
SLC12A6 的隐性突变与无脑回畸形的遗传性运动感觉神经病有关。2016 年报道了伴有 SLC12A6 杂合变体的早发性周围神经病患者。迄今为止,仅报道了五个家系和三个变体,其谱尚不清楚。在这里,我们旨在描述日本患者 SLC12A6 相关的 Charcot-Marie-Tooth(CMT)疾病的临床和突变谱。
我们从我们的 DNA 微阵列和靶向重测序数据中提取 SLC12A6 变体,这些数据是从日本神经科或神经儿科部门转介到我们遗传实验室的 2598 名临床疑似 CMT 患者中获得的。我们总结了这些患者的临床和遗传特征。
在七个不相关的家庭中,我们发现了一个先前报道的和三个新的可能致病的 SLC12A6 杂合变体,以及两个意义不确定的变体。这些患者的平均发病年龄为 17.5±16.1 岁。关于电生理学,运动神经传导速度的中位数为 39.6±9.5m/sec。我们首次观察到三名患者存在智力障碍。一名患者发生癫痫,其脑部 MRI 显示额叶和颞叶萎缩,而白质和胼胝体无变化。
无论 CMT 亚型如何,对于伴有中枢神经系统病变(如认知障碍和癫痫)的 CMT 患者,均应考虑筛查 SLC12A6 基因。