Division of Neurology, Department of Clinical Neurosciences, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada.
Divsion of Cardiology, Department of Cardiac Sciences, Libin Cardiovascular Institute, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada.
J Med Case Rep. 2022 Jun 25;16(1):248. doi: 10.1186/s13256-022-03437-0.
Hereditary transthyretin amyloidosis is an uncommon multisystem disorder caused by mutation of the transthyretin protein, leading to peripheral neuropathy often with autonomic features, cardiomyopathy, or a mixed phenotype. Multiple other organ systems can be involved with ophthalmologic, renal, hematologic, gastrointestinal, and/or genitourinary symptoms and signs. This often results in assessments by multiple specialists and significant delays before the diagnosis is recognized. With the recent advent of potentially lifesaving therapies, early diagnosis has become even more important. Our case highlights the protean aspects of this disease as well as the difficulty of making this diagnosis, especially in the absence of a clear family history.
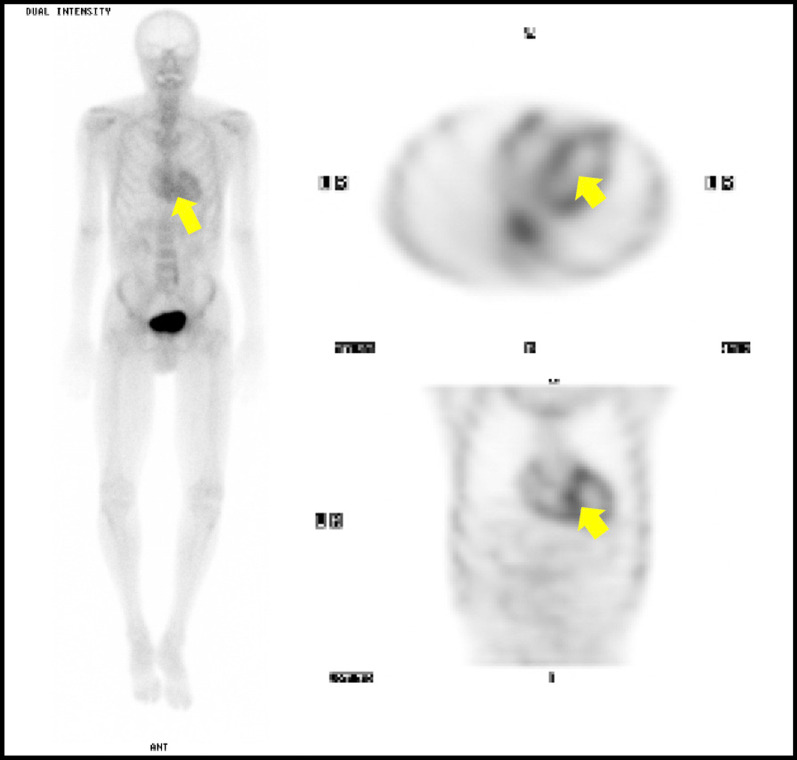
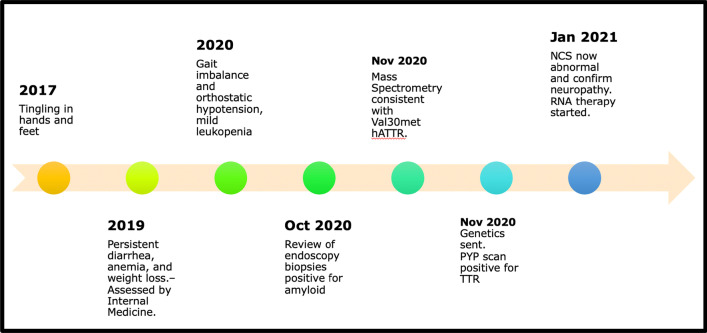
We report the case of a 64-year-old man of East-Asian descent who presented with diarrhea, mild anemia, and symptoms of peripheral neuropathy. Numerous investigations and specialist evaluations did not identify a cause. Progression of neurologic symptoms and the development of new hematologic abnormalities ultimately led to consideration of hereditary transthyretin amyloidosis. The diagnosis was confirmed after re-examining previously acquired gastrointestinal biopsies and pursuing genetic testing, which confirmed a pathogenic mutation in the transthyretin gene. He was subsequently started on a novel gene-silencing therapy. On clinical follow-up 8 months after initiation of therapy, the patient described stabilization of previously progressive numbness, weakness, and weight loss with an unchanged neurologic examination and stable repeat electrophysiologic testing.
Hereditary transthyretin amyloidosis is a challenging disease to recognize in early stages owing to its multisystem and nonspecific manifestations. Recent approval of novel therapies highlights the importance of early diagnosis before irreversible organ damage occurs.
遗传性转甲状腺素蛋白淀粉样变性是一种罕见的多系统疾病,由转甲状腺素蛋白的突变引起,常导致周围神经病变,伴有自主神经特征、心肌病或混合表型。多个其他器官系统可能会受到影响,出现眼科、肾脏、血液、胃肠道和/或泌尿生殖系统的症状和体征。这通常会导致多个专科医生进行评估,并在诊断得到确认之前出现显著的延迟。随着最近可能挽救生命的治疗方法的出现,早期诊断变得更加重要。我们的病例突出了这种疾病的多形性,以及在没有明确家族史的情况下做出这种诊断的困难。
我们报告了一例 64 岁的东亚裔男性,他因腹泻、轻度贫血和周围神经病变症状就诊。大量的检查和专科医生评估未能确定病因。神经症状的进展和新的血液学异常的出现最终导致考虑遗传性转甲状腺素蛋白淀粉样变性。在重新检查先前获得的胃肠道活检并进行基因检测后,诊断得到了确认,该检测证实了转甲状腺素蛋白基因的致病性突变。随后,他开始接受一种新型基因沉默治疗。在开始治疗后 8 个月的临床随访中,患者描述先前进行性麻木、无力和体重减轻的情况稳定,神经检查无变化,重复电生理检查稳定。
由于遗传性转甲状腺素蛋白淀粉样变性具有多系统和非特异性表现,因此在早期识别具有挑战性。最近批准的新型治疗方法强调了在不可逆器官损伤发生之前进行早期诊断的重要性。