Department of Physics and Astronomy, The University of British Columbia, Vancouver, BC V6T 1Z1, Canada.
Djavad Mowafaghian Centre for Brain Health, The University of British Columbia, Vancouver, BC V6T 2B5, Canada.
ACS Chem Neurosci. 2022 Aug 3;13(15):2261-2280. doi: 10.1021/acschemneuro.1c00567. Epub 2022 Jul 15.
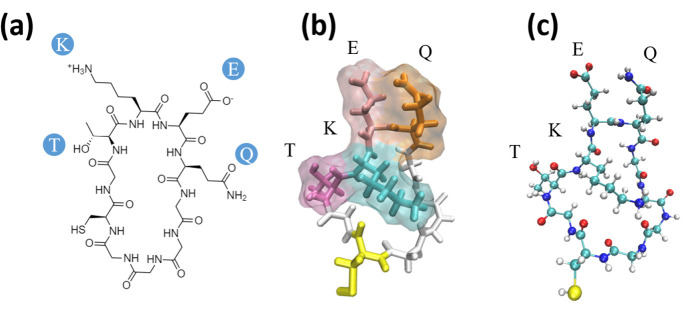
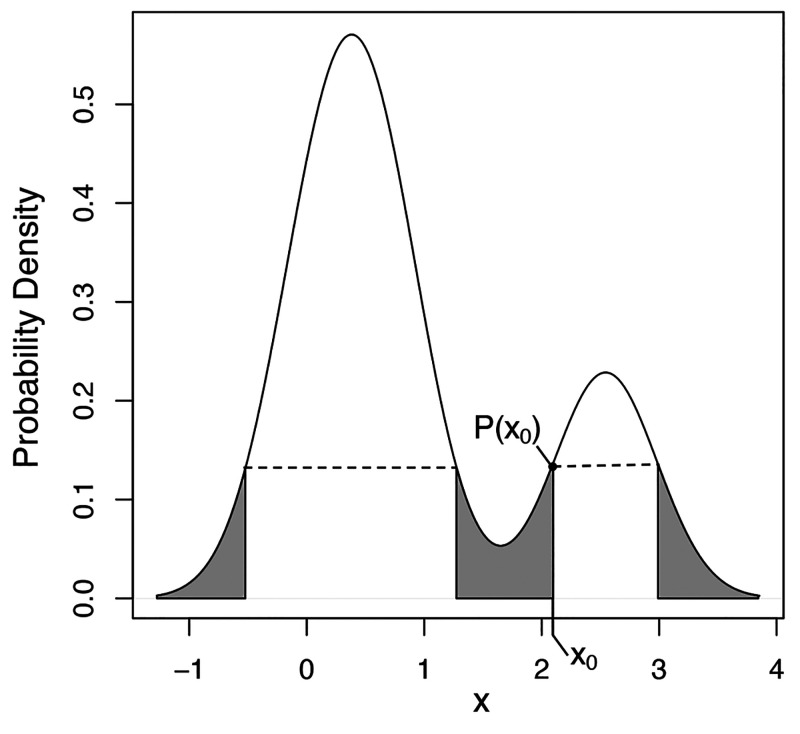
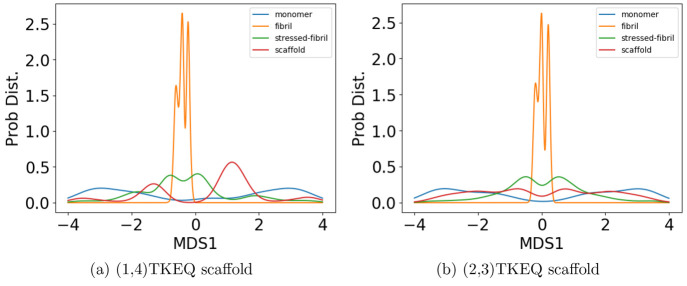
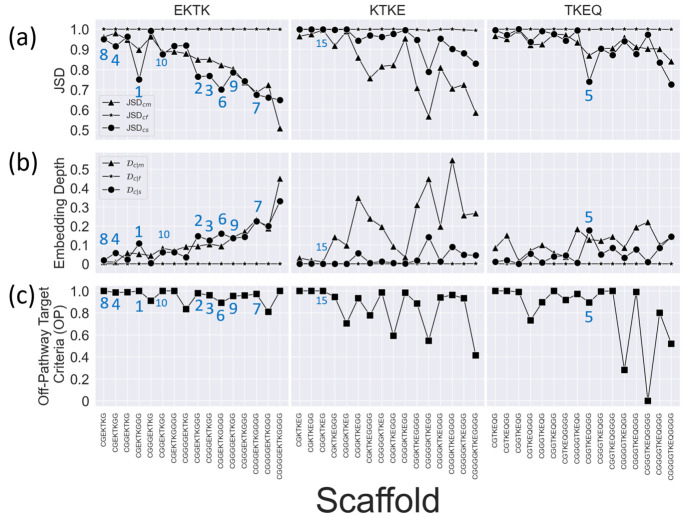
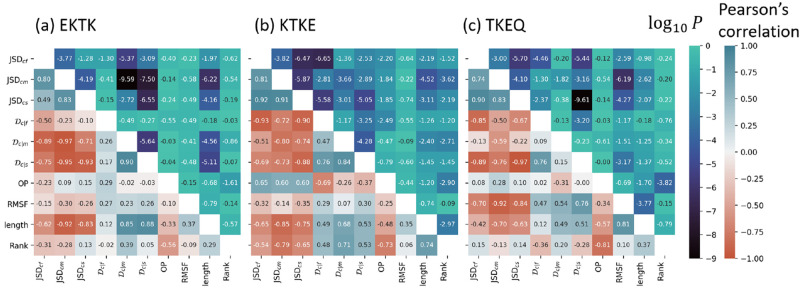
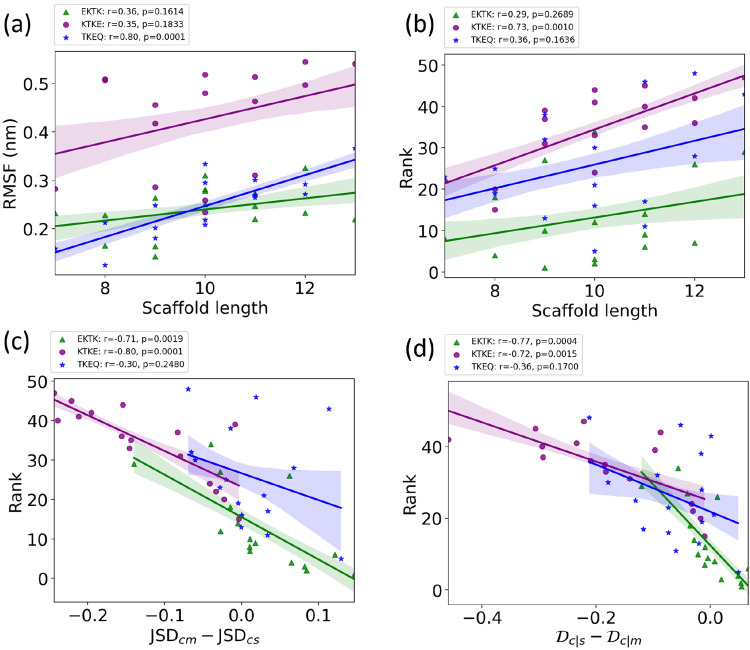
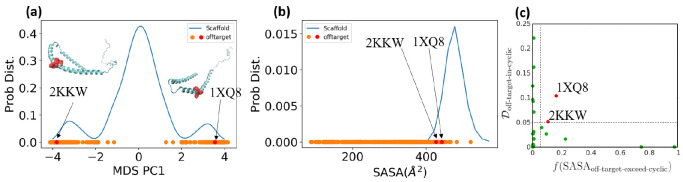
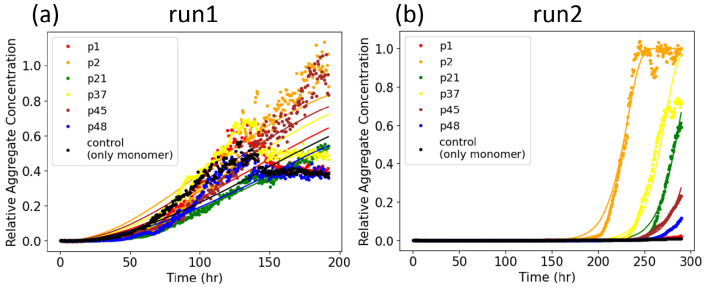
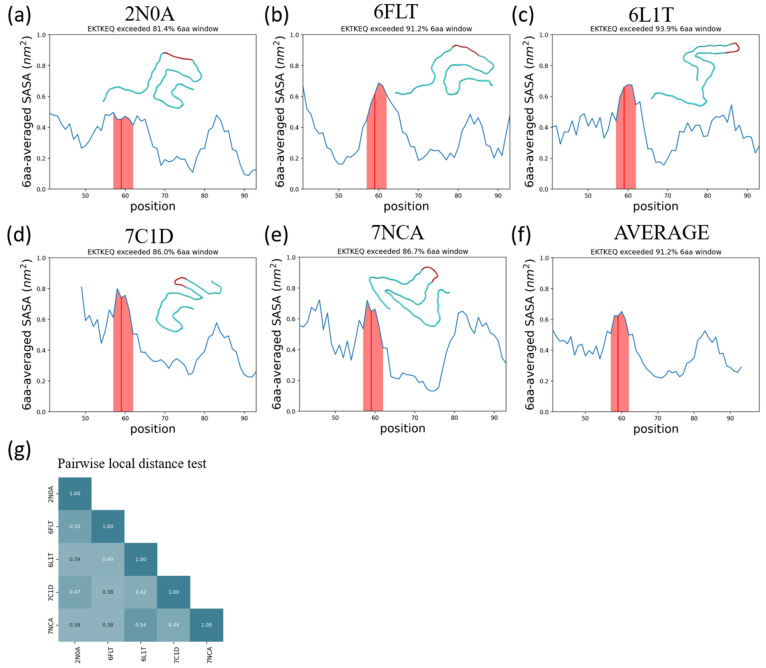
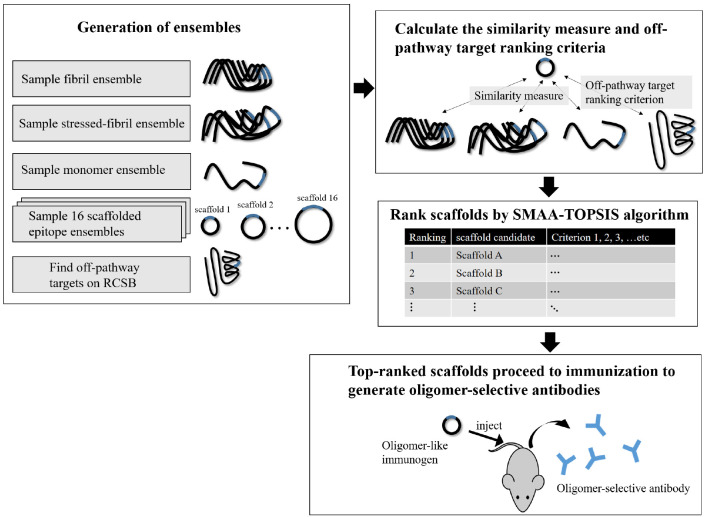
Effectively presenting epitopes on immunogens, in order to raise conformationally selective antibodies through active immunization, is a central problem in treating protein misfolding diseases, particularly neurodegenerative diseases such as Alzheimer's disease or Parkinson's disease. We seek to selectively target conformations enriched in toxic, oligomeric propagating species while sparing the healthy forms of the protein that are often more abundant. To this end, we computationally modeled scaffolded epitopes in cyclic peptides by inserting/deleting a variable number of flanking glycines ("glycindels") to best mimic a misfolding-specific conformation of an epitope of α-synuclein enriched in the oligomer ensemble, as characterized by a region most readily disordered and solvent-exposed in a stressed, partially denatured protofibril. We screen and rank the cyclic peptide scaffolds of α-synuclein based on their ensemble overlap properties with the fibril, oligomer-model and isolated monomer ensembles. We present experimental data of seeded aggregation that support nucleation rates consistent with computationally predicted cyclic peptide conformational similarity. We also introduce a method for screening against structured off-pathway targets in the human proteome by selecting scaffolds with minimal conformational similarity between their epitope and the same solvent-exposed primary sequence in structured human proteins. Different cyclic peptide scaffolds with variable numbers of glycines are predicted computationally to have markedly different conformational ensembles. Ensemble comparison and overlap were quantified by the Jensen-Shannon divergence and a new measure introduced here, the embedding depth, which determines the extent to which a given ensemble is subsumed by another ensemble and which may be a more useful measure in developing immunogens that confer conformational selectivity to an antibody.
有效地在免疫原上呈现表位,以通过主动免疫产生构象选择性抗体,是治疗蛋白质错误折叠疾病(尤其是阿尔茨海默病或帕金森病等神经退行性疾病)的核心问题。我们试图选择性地针对富含毒性、寡聚传播物种的构象,同时保留通常更为丰富的蛋白质的健康形式。为此,我们通过插入/删除可变数量的侧翼甘氨酸(“甘氨酸缺失”)来计算模拟环状肽中的支架表位,以最佳模拟富含寡聚体的α-突触核蛋白表位的特定错误折叠构象,该表位在处于压力下的部分变性原纤维中最容易无序和暴露于溶剂。我们基于与纤维、寡聚模型和分离单体的集合重叠特性对α-突触核蛋白的环状肽支架进行筛选和排序。我们提出了种子聚合的实验数据,这些数据支持与计算预测的环状肽构象相似性一致的成核速率。我们还引入了一种通过选择与结构人类蛋白质中暴露的相同原始序列的表位具有最小构象相似性的支架来筛选人类蛋白质组中结构偏离途径靶标的方法。具有可变数量甘氨酸的不同环状肽支架在计算上被预测具有明显不同的构象集合。通过杰恩斯-香农散度和此处引入的新度量——嵌入深度来量化集合比较和重叠,该度量确定了给定集合被另一个集合包含的程度,并且在开发赋予抗体构象选择性的免疫原时可能是更有用的度量。