Li Wei, Lu Zhe, Pan Dongqing, Zhang Zejian, He Hua, Wu Jiacheng, Peng Naixiong
Department of Urology, Shenzhen Longhua District Central Hospital, The Affiliated Central Hospital of Shenzhen Longhua District, Guangdong Medical University, Shenzhen, Guangdong Province, China.
Clinical Laboratory, Women and Children's Health Care Center of Hainan Province, Haikou, Hainan Province, China.
J Oncol. 2022 Jul 6;2022:5504173. doi: 10.1155/2022/5504173. eCollection 2022.
Tyrosine metabolism pathway-related genes were related to prostate cancer progression, which may be used as potential prognostic markers.
To dissect the dysregulation of tyrosine metabolism in prostate cancer and build a prognostic signature based on tyrosine metabolism-related genes for prostate cancer. . Cross-platform gene expression data of prostate cancer cohorts were collected from both The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO). Based on the expression of tyrosine metabolism-related enzymes (TMREs), an unsupervised consensus clustering method was used to classify prostate cancer patients into different molecular subtypes. We employed the least absolute shrinkage and selection operator (LASSO) Cox regression analysis to evaluate prognostic characteristics based on TMREs to obtain a prognostic effect. The nomogram model was established and used to synthesize molecular subtypes, prognostic characteristics, and clinicopathological features. Kaplan-Meier plots and logrank analysis were used to clarify survival differences between subtypes.
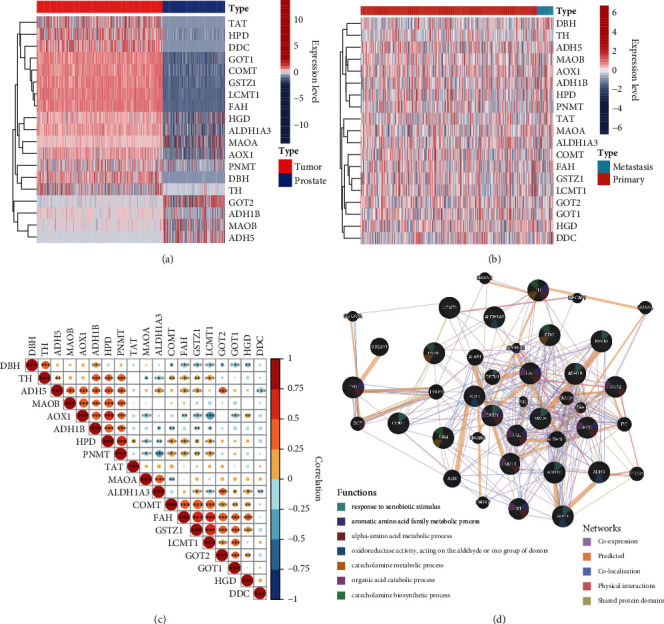
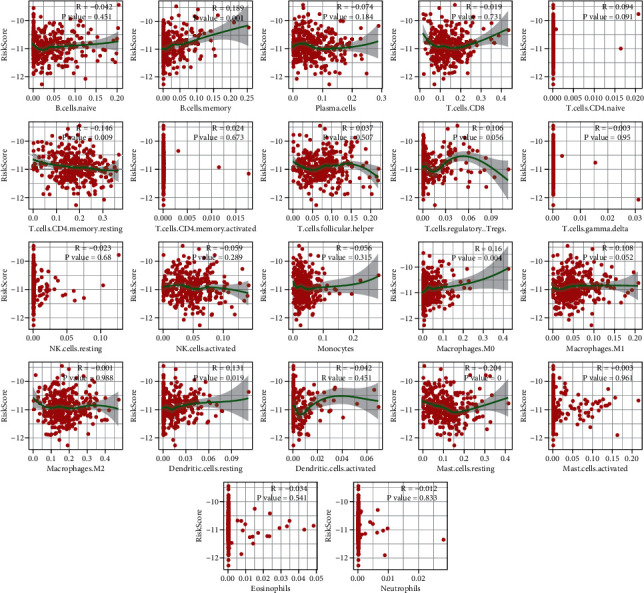
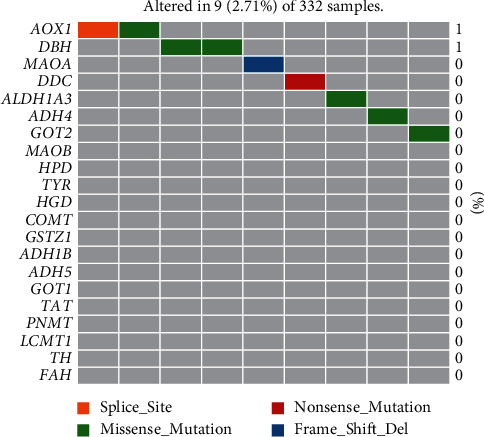
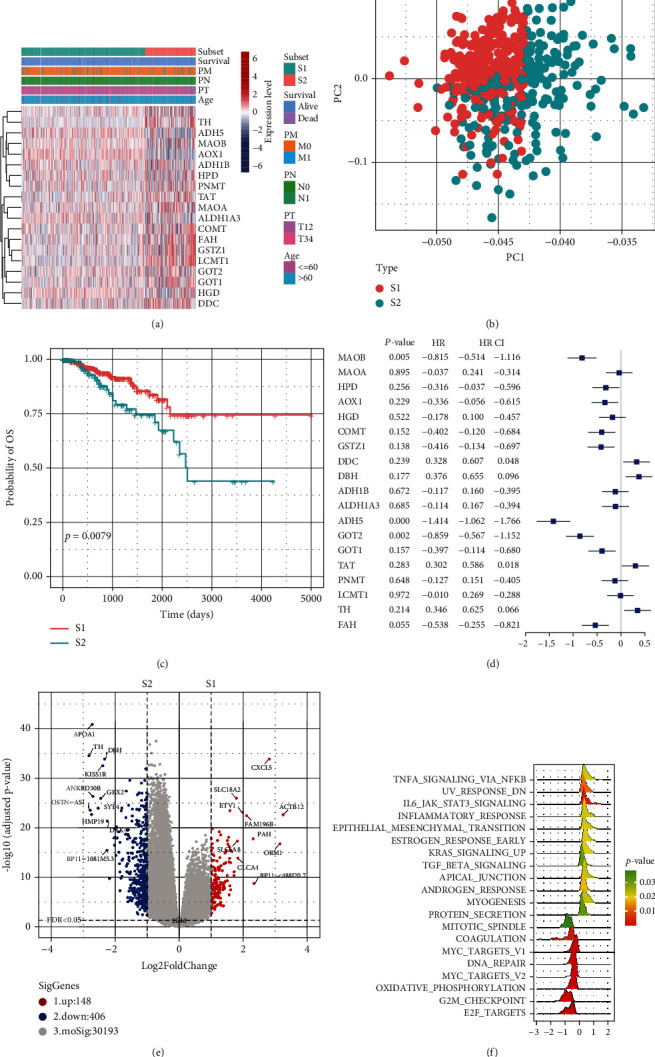
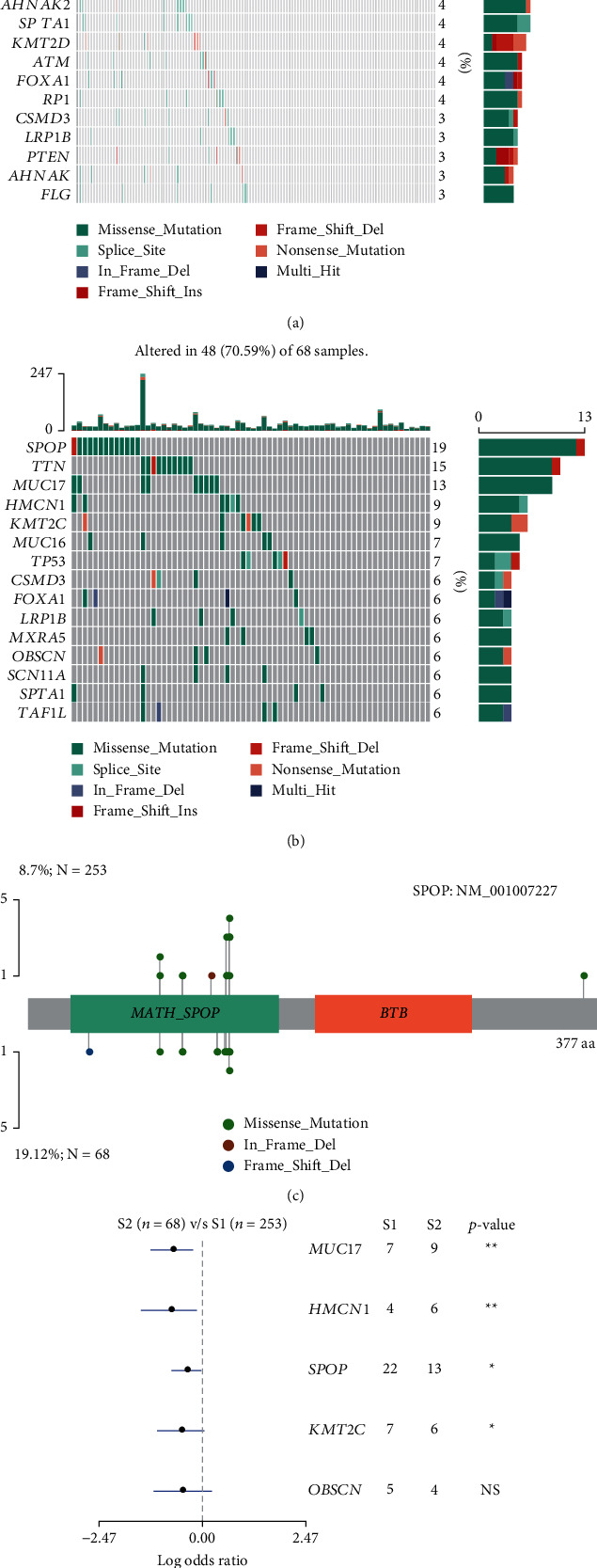
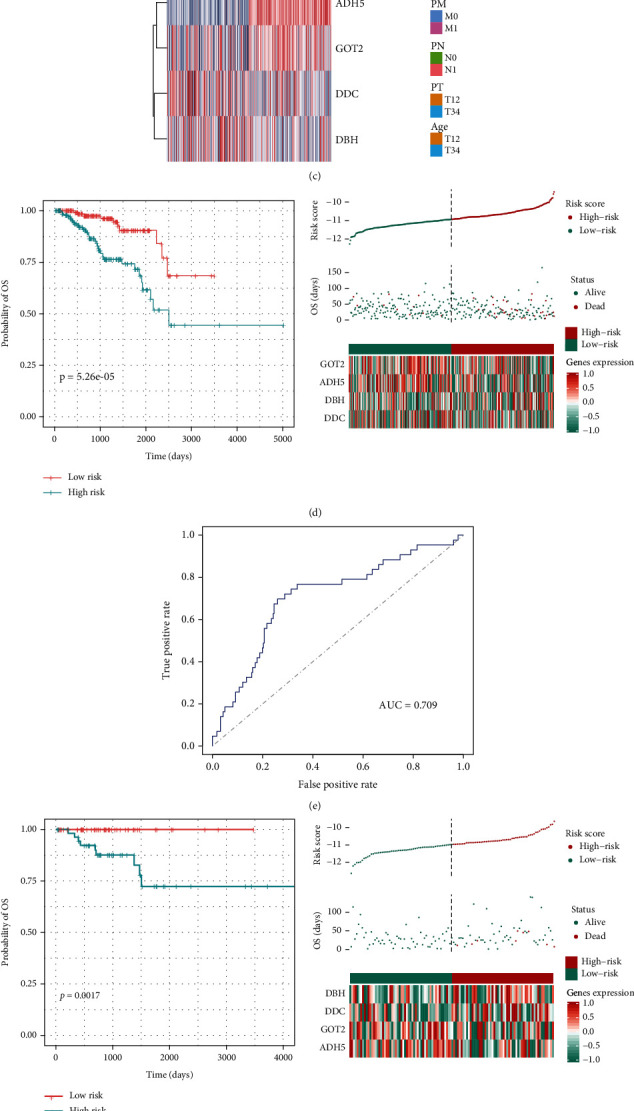
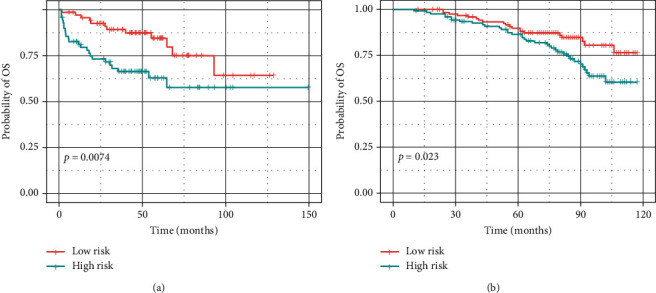
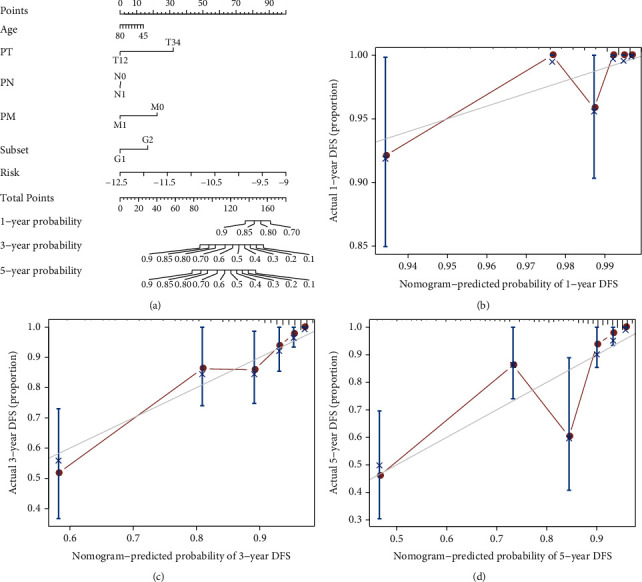
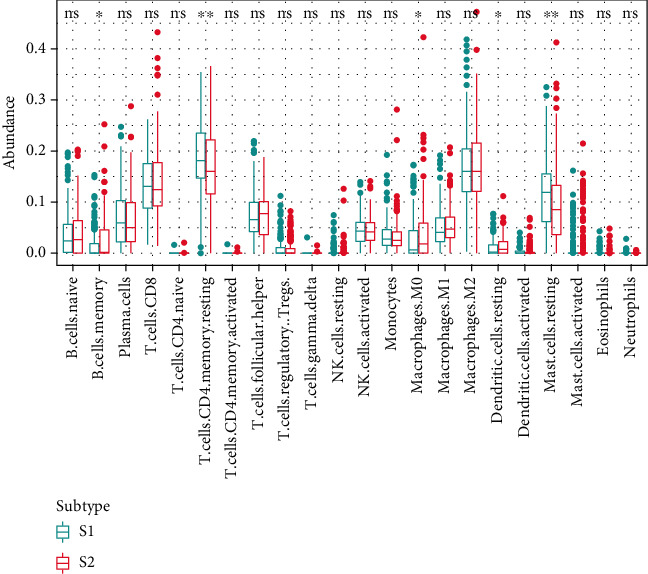
Based on the hierarchical clustering method and the expression profiles of TMREs, prostate cancer samples were assigned into two subgroups (S1, subgroup 1; S2, subgroup 2), and the Kaplan-Meier plot and logrank analysis showed distinct survival outcomes between S1 and S2 subgroups. We further established a four-gene-based prognostic signature, and both in-group testing dataset and out-group testing dataset indicated the robustness of this model. By combining the four gene-based signatures and clinicopathological features, the nomogram model achieved better survival outcomes than any single classifier. Interestingly, we found that immune-related pathways were significantly concentrated on S1-upregulated genes, and the abundance of memory B cells, CD4+ resting memory T cells, M0 macrophages, resting dendritic cells, and resting mast cells were significantly different between S1 and S2 subgroups.
Our results indicate the prognostic value of genes related to tyrosine metabolism in prostate cancer and provide inspiration for treatment and prevention strategies.
酪氨酸代谢途径相关基因与前列腺癌进展相关,可作为潜在的预后标志物。
剖析前列腺癌中酪氨酸代谢的失调情况,并基于酪氨酸代谢相关基因构建前列腺癌的预后特征模型。
从癌症基因组图谱(TCGA)和基因表达综合数据库(GEO)收集前列腺癌队列的跨平台基因表达数据。基于酪氨酸代谢相关酶(TMREs)的表达,采用无监督一致性聚类方法将前列腺癌患者分为不同的分子亚型。我们采用最小绝对收缩和选择算子(LASSO)Cox回归分析,基于TMREs评估预后特征以获得预后效果。建立列线图模型并用于综合分子亚型、预后特征和临床病理特征。采用Kaplan-Meier曲线和对数秩检验分析来阐明各亚型之间的生存差异。
基于层次聚类方法和TMREs的表达谱,前列腺癌样本被分为两个亚组(S1,亚组1;S2,亚组2),Kaplan-Meier曲线和对数秩检验分析显示S1和S2亚组之间有明显不同的生存结果。我们进一步建立了基于四个基因的预后特征模型,组内测试数据集和组外测试数据集均表明该模型具有稳健性。通过结合基于四个基因的特征模型和临床病理特征,列线图模型比任何单一分类器都能实现更好的生存结果。有趣的是,我们发现免疫相关途径显著集中于S1上调基因,且S1和S2亚组之间记忆B细胞、CD4 + 静息记忆T细胞、M0巨噬细胞、静息树突状细胞和静息肥大细胞的丰度存在显著差异。
我们的结果表明酪氨酸代谢相关基因在前列腺癌中的预后价值,并为治疗和预防策略提供了启示。