Department of Medical Genetics, College of Medicine, Umm Al-Qura University, P.O. Box 57543, Mecca, 21955, Saudi Arabia.
Department of Community Medicine, College of Medicine, Umm Al-Qura University, P.O. Box 57543, Mecca, 21955, Saudi Arabia.
Hum Genomics. 2022 Jul 19;16(1):22. doi: 10.1186/s40246-022-00398-9.
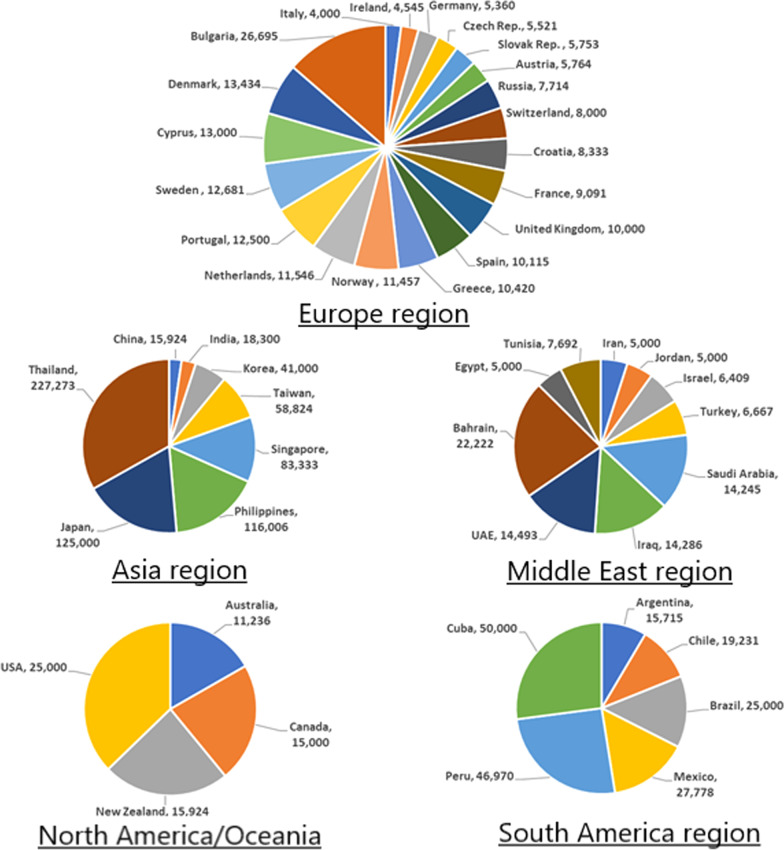
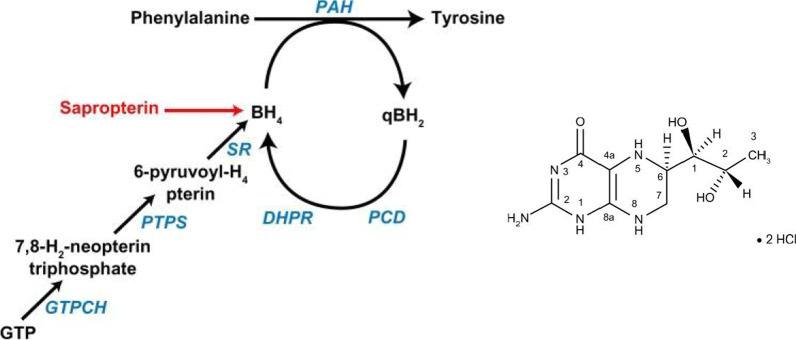
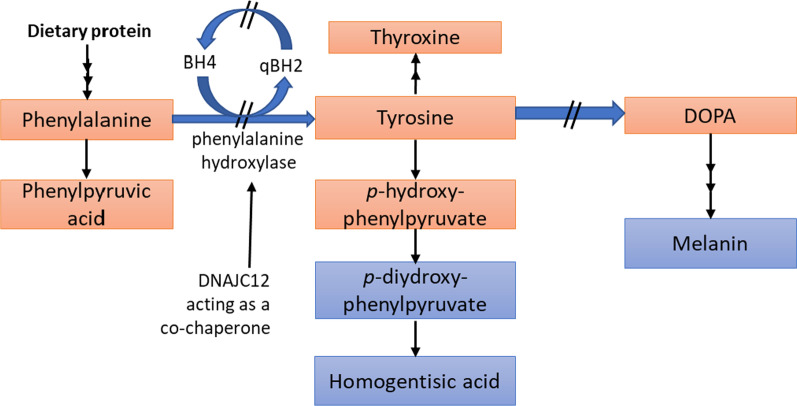
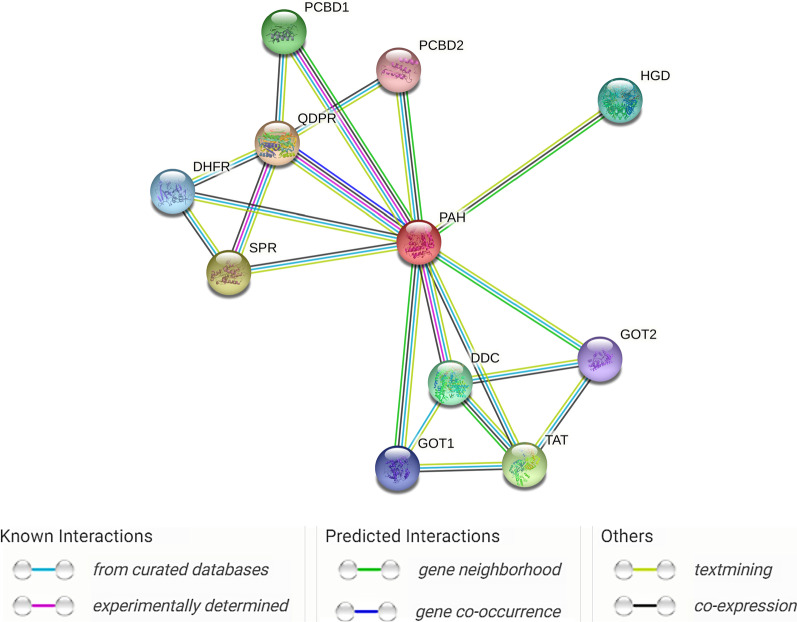
This review discusses the epidemiology, pathophysiology, genetic etiology, and management of phenylketonuria (PKU). PKU, an autosomal recessive disease, is an inborn error of phenylalanine (Phe) metabolism caused by pathogenic variants in the phenylalanine hydroxylase (PAH) gene. The prevalence of PKU varies widely among ethnicities and geographic regions, affecting approximately 1 in 24,000 individuals worldwide. Deficiency in the PAH enzyme or, in rare cases, the cofactor tetrahydrobiopterin results in high blood Phe concentrations, causing brain dysfunction. Untreated PKU, also known as PAH deficiency, results in severe and irreversible intellectual disability, epilepsy, behavioral disorders, and clinical features such as acquired microcephaly, seizures, psychological signs, and generalized hypopigmentation of skin (including hair and eyes). Severe phenotypes are classic PKU, and less severe forms of PAH deficiency are moderate PKU, mild PKU, mild hyperphenylalaninaemia (HPA), or benign HPA. Early diagnosis and intervention must start shortly after birth to prevent major cognitive and neurological effects. Dietary treatment, including natural protein restriction and Phe-free supplements, must be used to maintain blood Phe concentrations of 120-360 μmol/L throughout the life span. Additional treatments include the casein glycomacropeptide (GMP), which contains very limited aromatic amino acids and may improve immunological function, and large neutral amino acid (LNAA) supplementation to prevent plasma Phe transport into the brain. The synthetic BH4 analog, sapropterin hydrochloride (i.e., Kuvan®, BioMarin), is another potential treatment that activates residual PAH, thus decreasing Phe concentrations in the blood of PKU patients. Moreover, daily subcutaneous injection of pegylated Phe ammonia-lyase (i.e., pegvaliase; PALYNZIQ®, BioMarin) has promised gene therapy in recent clinical trials, and mRNA approaches are also being studied.
本文综述了苯丙酮尿症(PKU)的流行病学、病理生理学、遗传病因学和治疗方法。PKU 是一种常染色体隐性遗传疾病,由苯丙氨酸羟化酶(PAH)基因突变引起,是苯丙氨酸代谢的先天缺陷。PKU 在不同种族和地理区域的患病率差异很大,全球约有 1/24,000 的人受其影响。PAH 酶缺乏或在极少数情况下四氢生物蝶呤(BH4)辅因子缺乏会导致血液中苯丙氨酸浓度升高,从而导致大脑功能障碍。未经治疗的 PKU 也称为 PAH 缺乏症,会导致严重且不可逆转的智力残疾、癫痫、行为障碍以及获得性小头畸形、癫痫发作、心理迹象和皮肤(包括头发和眼睛)普遍色素减退等临床特征。严重表型为经典 PKU,PAH 缺乏症的较轻形式为中度 PKU、轻度 PKU、轻度高苯丙氨酸血症(HPA)或良性 HPA。早期诊断和干预必须在出生后不久开始,以防止对认知和神经功能产生重大影响。饮食治疗包括天然蛋白质限制和无苯丙氨酸补充剂,以维持整个生命周期内血液苯丙氨酸浓度在 120-360μmol/L 之间。其他治疗方法包括含有非常有限芳香族氨基酸的酪蛋白糖巨肽(GMP),可能改善免疫功能,以及大中性氨基酸(LNAA)补充剂,以防止血液中的苯丙氨酸转运到大脑。合成 BH4 类似物盐酸沙丙蝶呤(即 Kuvan®,BioMarin)是另一种潜在的治疗方法,可激活残留的 PAH,从而降低 PKU 患者血液中的苯丙氨酸浓度。此外,聚乙二醇化苯丙氨酸氨解酶(即 pegvaliase;PALYNZIQ®,BioMarin)的每日皮下注射在最近的临床试验中有望实现基因治疗,mRNA 方法也正在研究中。