Villa Metabolica, University Medical Center Mainz, Mainz, Germany.
Rare Disease Research Partners, Amersham, UK.
Orphanet J Rare Dis. 2022 Jul 23;17(1):287. doi: 10.1186/s13023-022-02422-6.
Alpha-mannosidosis is a rare autosomal recessive lysosomal storage disorder (LSD) caused by reduced activity of alpha-mannosidase. Clinical manifestations include skeletal dysmorphism, mental impairment, hearing loss and recurrent infections. The severe type of the disease leads to early childhood death, while patients with milder forms can live into adulthood. There are no mortality studies to date. This study aimed to investigate the age at death and the causes of death of patients with alpha-mannosidosis who had not received disease-modifying treatment.
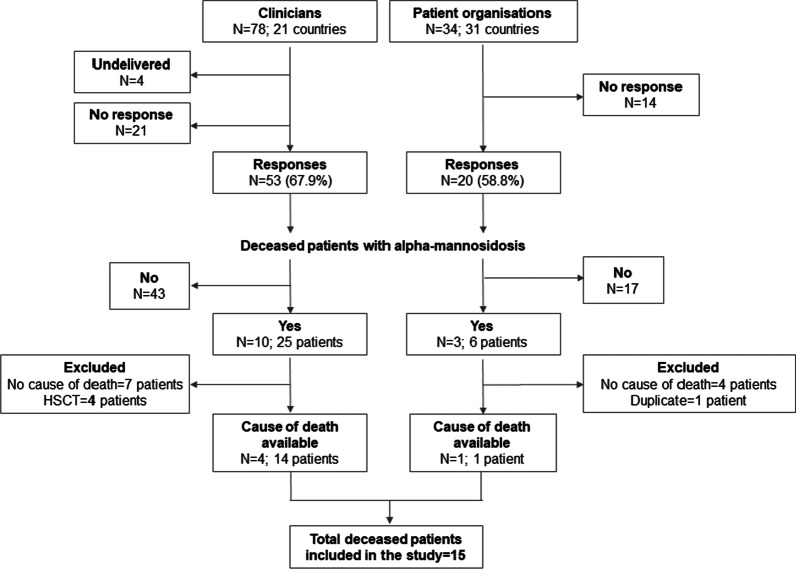
Clinicians and LSD patient organisations (POs) from 33 countries were invited to complete a questionnaire between April-May 2021. Cause of death and age at death was available for 15 patients. A literature review identified seven deceased patients that met the inclusion criteria.
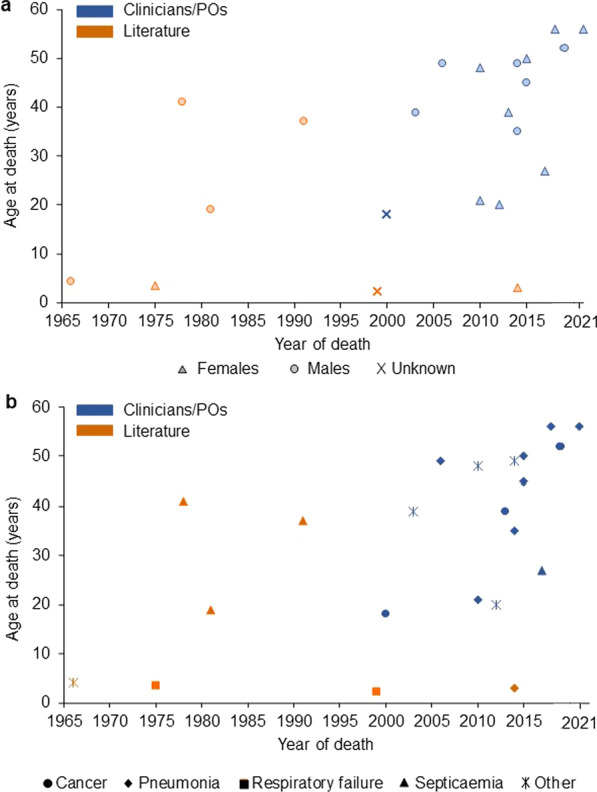
Median age at death for patients reported by clinicians/POs was 45 years (mean 40.3 ± 13.2, range 18-56, n = 15); 53% were female. One death occurred during the patient's second decade of life, and 14 out of 15 deaths (93.3%) during or after the patients' third decade, including four (26.7%) during their sixth decade. Median age at death for patients identified from the literature was 4.3 years (mean 15.7 ± 17.0, range 2.2-41, n = 7); two were female. Four of the seven patients (57.1%) died within the first decade of life. Seven of 15 deaths (46.7%) reported by clinicians/POs were recorded as pneumonia and three (20.0%) as cancer. Other causes of death included acute renal failure due to sepsis after intestinal perforation, decrease of red blood cells of unknown origin, kidney failure with systemic lupus erythematosus, aortic valve insufficiency leading to heart failure, and dehydration due to catatonia. Three out of seven causes of death (42.9%) reported in the literature were associated with septicaemia, two (28.6%) with respiratory failure and one to pneumonia following aspiration.
This study suggests that pneumonia has been the primary cause of death during recent decades in untreated patients with alpha-mannosidosis, followed by cancer. Determining the causes of mortality and life expectancy in these patients is crucial to further improve our understanding of the natural history of alpha-mannosidosis.
α-甘露糖苷贮积症是一种罕见的常染色体隐性溶酶体贮积病(LSD),由α-甘露糖苷酶活性降低引起。临床表现包括骨骼发育不良、智力障碍、听力损失和反复感染。该疾病的严重类型导致儿童早期死亡,而轻度形式的患者可存活至成年。迄今为止,尚无关于死亡率的研究。本研究旨在调查未接受疾病修正治疗的α-甘露糖苷贮积症患者的死亡年龄和死因。
邀请来自 33 个国家的临床医生和 LSD 患者组织(PO)于 2021 年 4 月至 5 月之间填写问卷。临床医生/PO 提供了 15 名患者的死亡原因和死亡年龄。文献综述确定了符合纳入标准的七名已故患者。
临床医生/PO 报告的患者的中位死亡年龄为 45 岁(平均 40.3±13.2,范围 18-56,n=15);53%为女性。有 1 例死亡发生在患者的第二个十年,15 例死亡中有 14 例(93.3%)发生在患者的第三个十年或之后,包括 4 例(26.7%)发生在第六个十年。文献中确定的患者的中位死亡年龄为 4.3 岁(平均 15.7±17.0,范围 2.2-41,n=7);2 名患者为女性。7 名患者中有 4 名(57.1%)在生命的第一个十年内死亡。临床医生/PO 报告的 15 例死亡中有 7 例(46.7%)记录为肺炎,3 例(20.0%)为癌症。其他死因包括肠穿孔后脓毒症引起的急性肾衰竭、不明原因的红细胞减少、伴有系统性红斑狼疮的肾衰竭、主动脉瓣关闭不全导致心力衰竭以及因紧张性木僵导致的脱水。文献中报告的 7 例死因中有 3 例(42.9%)与败血症有关,2 例(28.6%)与呼吸衰竭有关,1 例与吸入性肺炎有关。
本研究表明,肺炎是近年来未经治疗的α-甘露糖苷贮积症患者死亡的主要原因,其次是癌症。确定这些患者的死亡率和预期寿命的原因对于进一步了解α-甘露糖苷贮积症的自然史至关重要。