Malm Dag, Nilssen Øivind
Department of Gastroenterology, University Hospital of North Norway, NO-9038, Norway.
Orphanet J Rare Dis. 2008 Jul 23;3:21. doi: 10.1186/1750-1172-3-21.
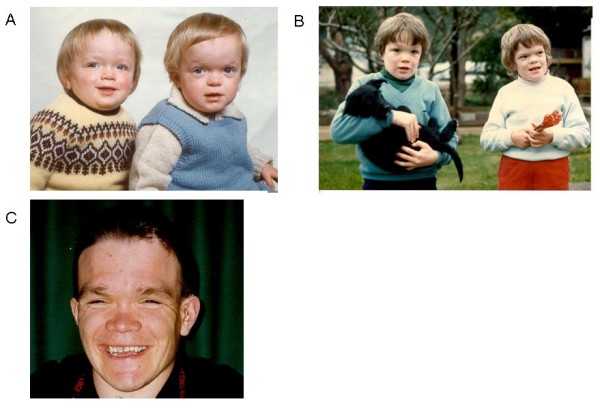
Alpha-mannosidosis is an inherited lysosomal storage disorder characterized by immune deficiency, facial and skeletal abnormalities, hearing impairment, and intellectual disability. It occurs in approximately 1 of 500,000 live births. The children are often born apparently normal, and their condition worsens progressively. Some children are born with ankle equinus or develop hydrocephalus in the first year of life. Main features are immune deficiency (manifested by recurrent infections, especially in the first decade of life), skeletal abnormalities (mild-to-moderate dysostosis multiplex, scoliosis and deformation of the sternum), hearing impairment (moderate-to-severe sensorineural hearing loss), gradual impairment of mental functions and speech, and often, periods of psychosis. Associated motor function disturbances include muscular weakness, joint abnormalities and ataxia. The facial trait include large head with prominent forehead, rounded eyebrows, flattened nasal bridge, macroglossia, widely spaced teeth, and prognathism. Slight strabismus is common. The clinical variability is significant, representing a continuum in severity. The disorder is caused by lysosomal alpha-mannosidase deficiency. Alpha-mannosidosis is inherited in an autosomal recessive fashion and is caused by mutations in the MAN2B1 gene located on chromosome 19 (19 p13.2-q12). Diagnosis is made by measuring acid alpha-mannosidase activity in leukocytes or other nucleated cells and can be confirmed by genetic testing. Elevated urinary secretion of mannose-rich oligosaccharides is suggestive, but not diagnostic. Differential diagnoses are mainly the other lysosomal storage diseases like the mucopolysaccharidoses. Genetic counseling should be given to explain the nature of the disease and to detect carriers. Antenatal diagnosis is possible, based on both biochemical and genetic methods. The management should be pro-active, preventing complications and treating manifestations. Infections must be treated frequently. Otolaryngological treatment of fluid in the middle ear is often required and use of hearing aids is invariably required. Early educational intervention for development of social skills is needed and physiotherapy is important to improve bodily function. Orthopedic surgery may be necessary. The long-term prognosis is poor. There is an insidiously slow progression of neuromuscular and skeletal deterioration over several decades, making most patients wheel-chair dependent. No patients manage to be completely socially independent. Many patients are over 50 years of age.
α-甘露糖苷贮积症是一种遗传性溶酶体贮积病,其特征为免疫缺陷、面部和骨骼异常、听力障碍及智力残疾。该病在每50万例活产中约有1例发生。患儿出生时通常外表正常,但其病情会逐渐恶化。一些患儿出生时即有马蹄足畸形,或在出生后第一年出现脑积水。主要特征包括免疫缺陷(表现为反复感染,尤其是在生命的第一个十年)、骨骼异常(轻度至中度多发性骨发育不良、脊柱侧弯和胸骨变形)、听力障碍(中度至重度感音神经性听力损失)、精神功能和言语逐渐受损,且常伴有精神病发作期。相关的运动功能障碍包括肌肉无力、关节异常和共济失调。面部特征包括头大、前额突出、眉毛圆润、鼻梁扁平、巨舌、牙齿间距宽和下颌前突。轻度斜视很常见。临床变异性很大,严重程度呈连续变化。该疾病由溶酶体α-甘露糖苷酶缺乏引起。α-甘露糖苷贮积症以常染色体隐性方式遗传,由位于19号染色体(19p13.2 - q12)上的MAN2B1基因突变所致。通过检测白细胞或其他有核细胞中的酸性α-甘露糖苷酶活性进行诊断,可通过基因检测予以确诊。尿中富含甘露糖的寡糖分泌增加有提示作用,但不能确诊。鉴别诊断主要是其他溶酶体贮积病,如黏多糖贮积症。应提供遗传咨询以解释疾病的性质并检测携带者。基于生化和基因方法可进行产前诊断。治疗应积极主动,预防并发症并治疗症状表现。必须经常治疗感染。中耳积液通常需要进行耳鼻喉科治疗,且总是需要使用助听器。需要早期进行社交技能发展的教育干预,物理治疗对于改善身体功能很重要。可能需要进行矫形手术。长期预后较差。在几十年间,神经肌肉和骨骼会逐渐缓慢恶化,导致大多数患者依赖轮椅。没有患者能够完全实现社会独立。许多患者年龄超过50岁。