Ng Rachel H, Lee Jihoon W, Baloni Priyanka, Diener Christian, Heath James R, Su Yapeng
Institute for Systems Biology, Seattle, WA, United States.
Department of Bioengineering, University of Washington, Seattle, WA, United States.
Front Oncol. 2022 Jul 7;12:914594. doi: 10.3389/fonc.2022.914594. eCollection 2022.
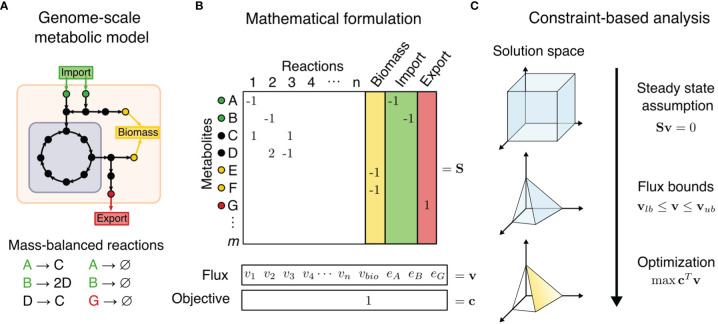
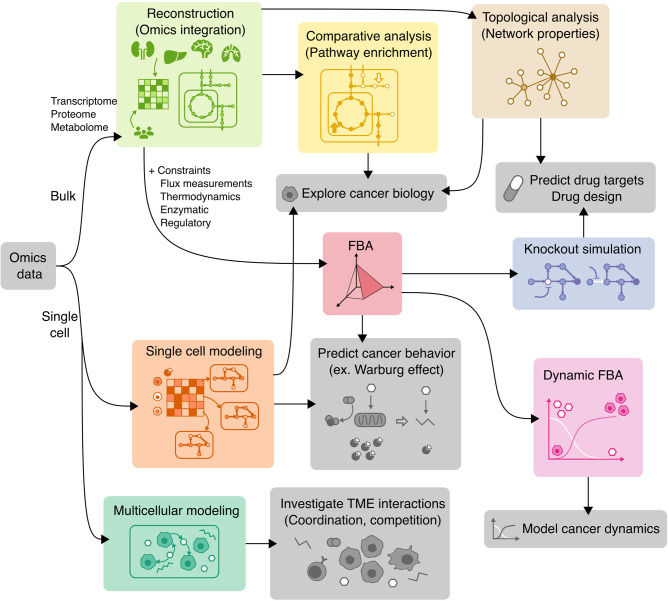
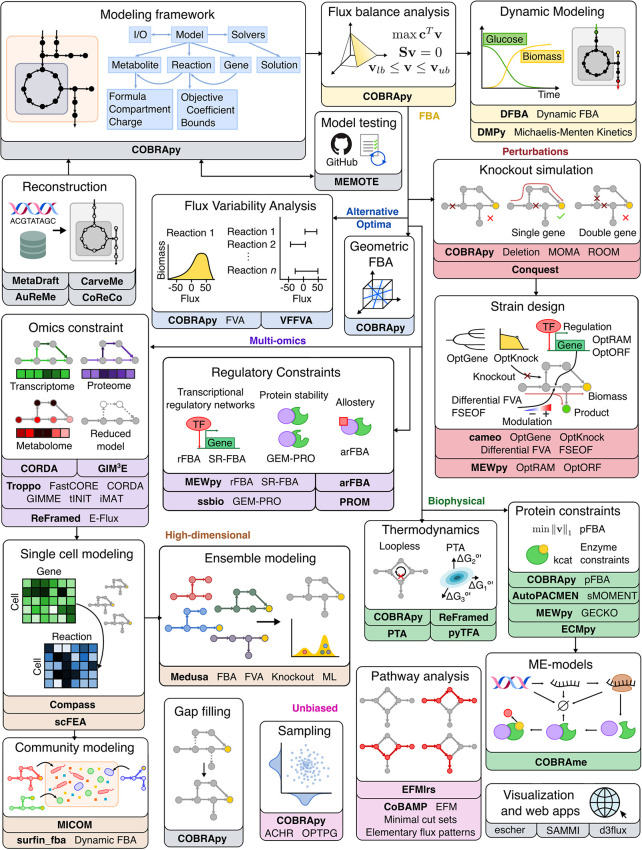
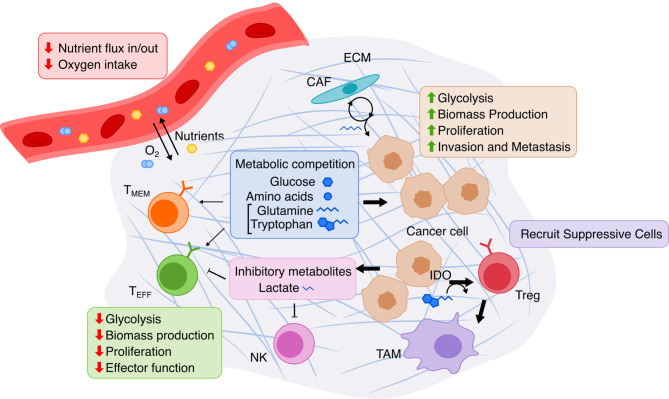
The influence of metabolism on signaling, epigenetic markers, and transcription is highly complex yet important for understanding cancer physiology. Despite the development of high-resolution multi-omics technologies, it is difficult to infer metabolic activity from these indirect measurements. Fortunately, genome-scale metabolic models and constraint-based modeling provide a systems biology framework to investigate the metabolic states and define the genotype-phenotype associations by integrations of multi-omics data. Constraint-Based Reconstruction and Analysis (COBRA) methods are used to build and simulate metabolic networks using mathematical representations of biochemical reactions, gene-protein reaction associations, and physiological and biochemical constraints. These methods have led to advancements in metabolic reconstruction, network analysis, perturbation studies as well as prediction of metabolic state. Most computational tools for performing these analyses are written for MATLAB, a proprietary software. In order to increase accessibility and handle more complex datasets and models, community efforts have started to develop similar open-source tools in Python. To date there is a comprehensive set of tools in Python to perform various flux analyses and visualizations; however, there are still missing algorithms in some key areas. This review summarizes the availability of Python software for several components of COBRA methods and their applications in cancer metabolism. These tools are evolving rapidly and should offer a readily accessible, versatile way to model the intricacies of cancer metabolism for identifying cancer-specific metabolic features that constitute potential drug targets.
代谢对信号传导、表观遗传标记和转录的影响极为复杂,但对于理解癌症生理学却很重要。尽管高分辨率多组学技术不断发展,但从这些间接测量中推断代谢活性仍很困难。幸运的是,基因组规模的代谢模型和基于约束的建模提供了一个系统生物学框架,可通过整合多组学数据来研究代谢状态并定义基因型-表型关联。基于约束的重建与分析(COBRA)方法用于使用生化反应、基因-蛋白质反应关联以及生理和生化约束的数学表示来构建和模拟代谢网络。这些方法推动了代谢重建、网络分析、扰动研究以及代谢状态预测等方面的进展。大多数用于执行这些分析的计算工具是为专有软件MATLAB编写的。为了提高可及性并处理更复杂的数据集和模型,社区已开始在Python中开发类似的开源工具。到目前为止,Python中有一套全面的工具可用于执行各种通量分析和可视化;然而,在一些关键领域仍然缺少算法。这篇综述总结了用于COBRA方法几个组件的Python软件的可用性及其在癌症代谢中的应用。这些工具正在迅速发展,应该能提供一种易于获取、通用的方式来模拟癌症代谢的复杂性,以识别构成潜在药物靶点的癌症特异性代谢特征。