Center for Vascular and Inflammatory Diseases, University of Maryland School of Medicine, Baltimore, Maryland, United States.
Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, Maryland, United States.
Thromb Haemost. 2022 Nov;122(11):1858-1868. doi: 10.1055/a-1910-4538. Epub 2022 Jul 27.
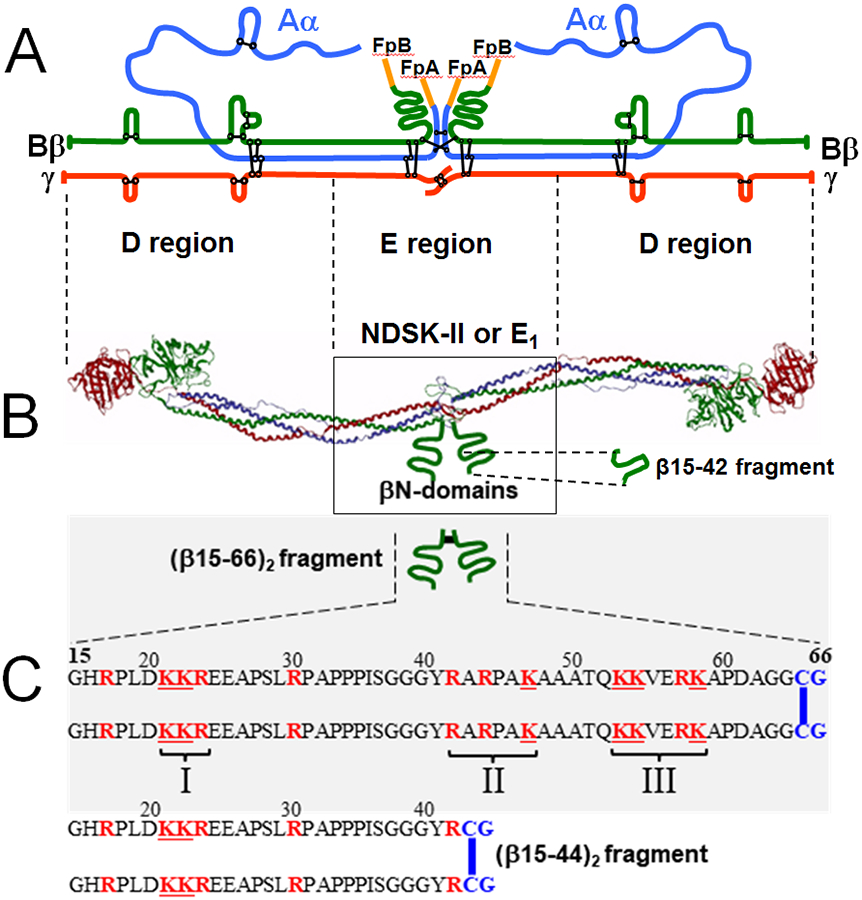
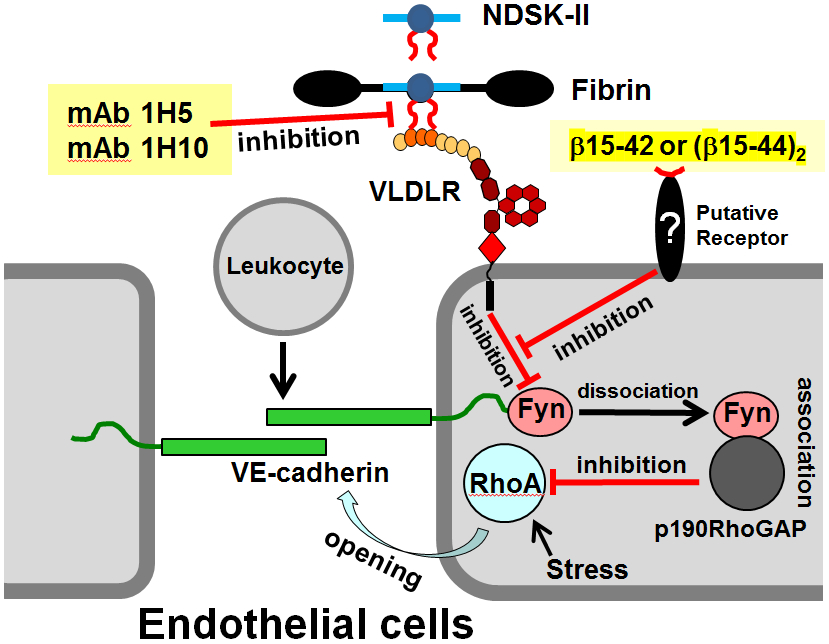
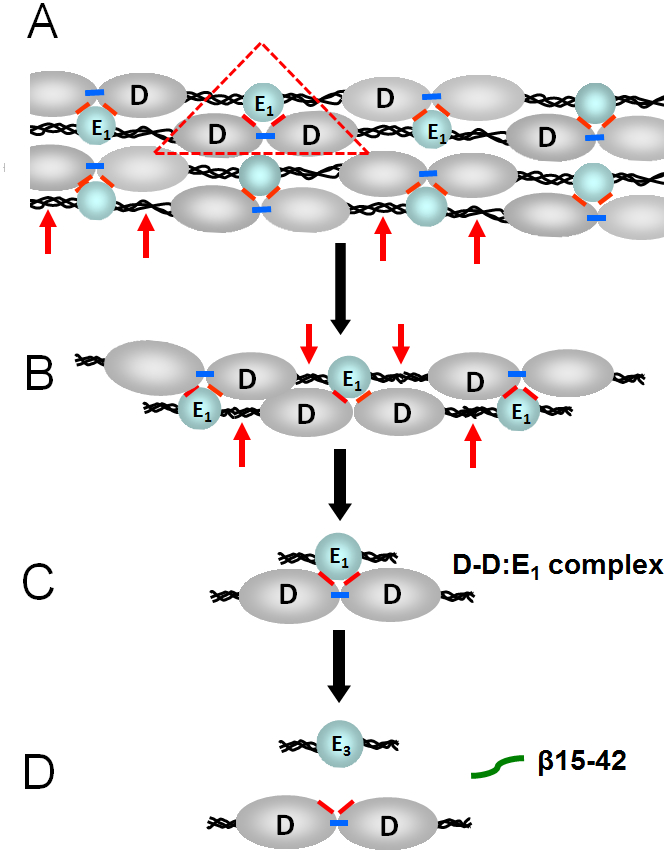
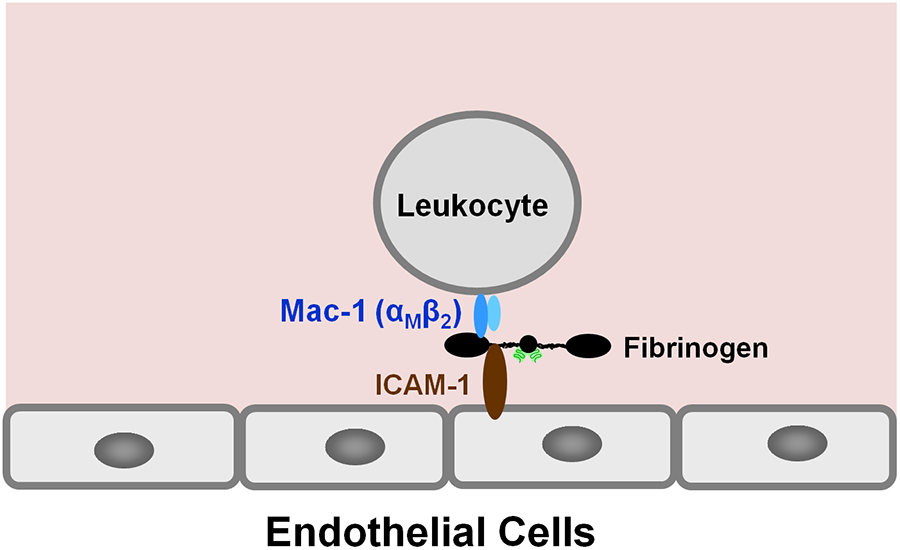
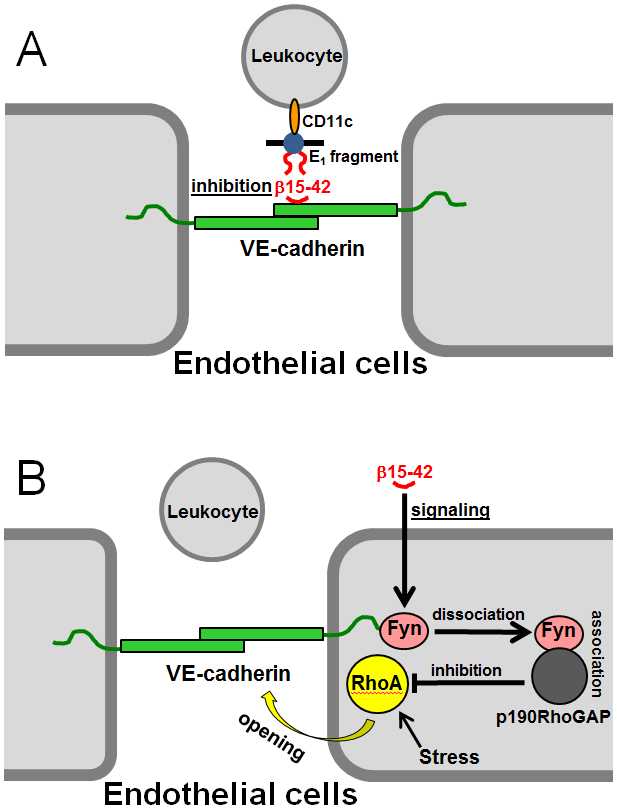
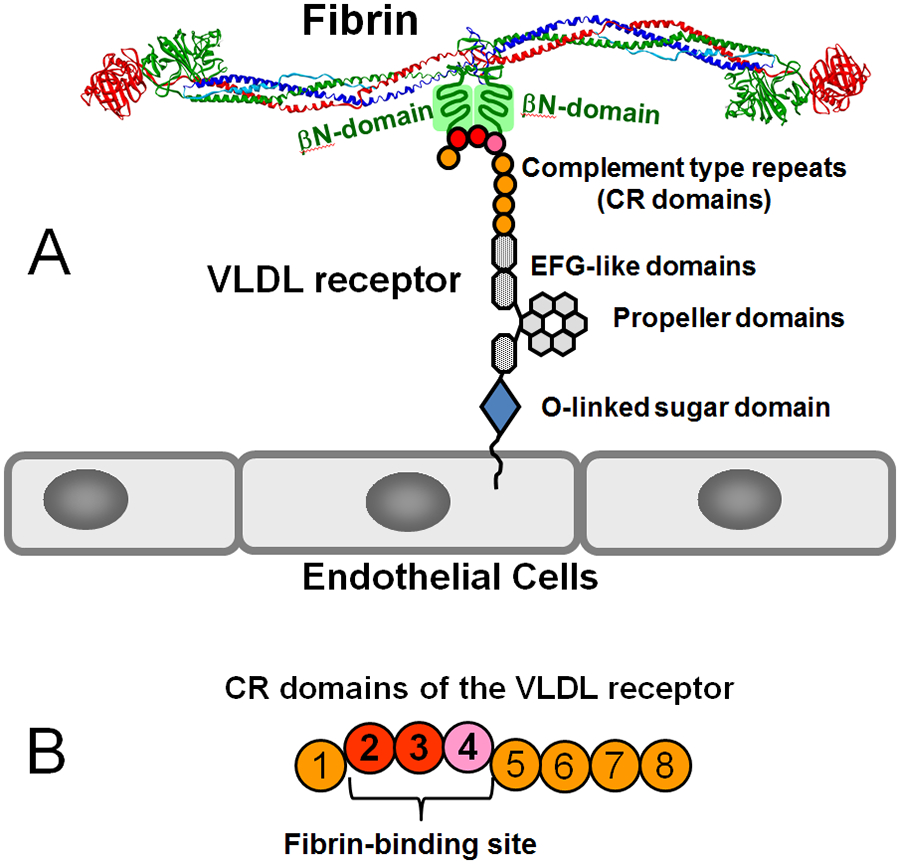
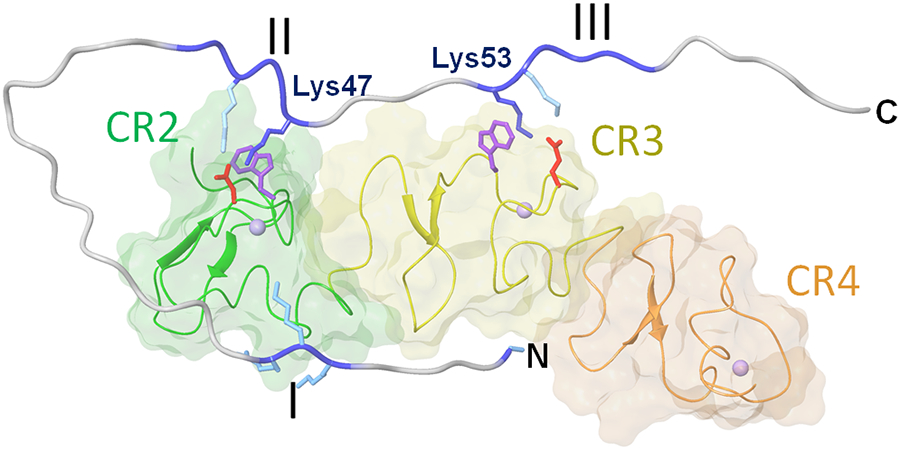
Numerous studies have revealed the involvement of fibrinogen in the inflammatory response. To explain the molecular mechanisms underlying fibrinogen-dependent inflammation, two bridging mechanisms have been proposed in which fibrin(ogen) bridges leukocytes to endothelial cells. The first mechanism suggests that bridging occurs via the interaction of fibrinogen with the leukocyte receptor Mac-1 and the endothelial receptor ICAM-1 (intercellular adhesion molecule-1), which promotes leukocyte transmigration and enhances inflammation. The second mechanism includes bridging of leukocytes to the endothelium by fibrin degradation product E fragment through its interaction with leukocyte receptor CD11c and endothelial VE-cadherin to promote leukocyte transmigration. The role of E in promoting inflammation is inhibited by the fibrin-derived β15-42 fragment, and this has been suggested to result from its ability to compete for the E-VE-cadherin interaction and to trigger signaling pathways through the src kinase Fyn. Our recent study revealed that the β15-42 fragment is ineffective in inhibiting the E- or fibrin-VE-cadherin interaction, leaving the proposed signaling mechanism as the only viable explanation for the inhibitory function of β15-42. We have discovered that fibrin interacts with the very-low-density lipoprotein (VLDL) receptor, and this interaction triggers a signaling pathway that promotes leukocyte transmigration through inhibition of the src kinase Fyn. This pathway is inhibited by another pathway induced by the interaction of β15-42 with a putative endothelial receptor. In this review, we briefly describe the previously proposed molecular mechanisms underlying fibrin-dependent inflammation and their advantages/disadvantages and summarize our recent studies of the novel VLDL receptor-dependent pathway of leukocyte transmigration which plays an important role in fibrin-dependent inflammation.
许多研究已经揭示了纤维蛋白原在炎症反应中的作用。为了解释纤维蛋白原依赖性炎症的分子机制,提出了两种桥接机制,其中纤维蛋白原将白细胞与内皮细胞桥接。第一种机制表明,桥接通过纤维蛋白原与白细胞受体 Mac-1 和内皮细胞受体 ICAM-1(细胞间黏附分子-1)的相互作用发生,促进白细胞迁移并增强炎症。第二种机制包括纤维蛋白降解产物 E 片段通过与其白细胞受体 CD11c 和内皮 VE-钙黏蛋白相互作用,将白细胞桥接到内皮细胞上,从而促进白细胞迁移。E 片段促进炎症的作用被纤维蛋白衍生的 β15-42 片段抑制,这被认为是由于其能够竞争 E-VE-钙黏蛋白相互作用并通过 src 激酶 Fyn 触发信号通路所致。我们最近的研究表明,β15-42 片段在抑制 E 或纤维蛋白-VE-钙黏蛋白相互作用方面无效,这使得提出的信号机制成为 β15-42 抑制功能的唯一可行解释。我们发现纤维蛋白与极低密度脂蛋白 (VLDL) 受体相互作用,这种相互作用触发了一种信号通路,通过抑制 src 激酶 Fyn 促进白细胞迁移。该通路被 β15-42 与假定的内皮受体相互作用诱导的另一条通路抑制。在这篇综述中,我们简要描述了以前提出的纤维蛋白依赖性炎症的分子机制及其优缺点,并总结了我们最近关于白细胞迁移的新型 VLDL 受体依赖性通路的研究,该通路在纤维蛋白依赖性炎症中发挥重要作用。