Department of Physiology, University of Texas Health Science Center at San Antonio, San Antonio, Texas.
Division of Nephrology, Department of Medicine, Stanford University School of Medicine, Palo Alto, California.
Am J Physiol Renal Physiol. 2022 Oct 1;323(4):F468-F478. doi: 10.1152/ajprenal.00384.2021. Epub 2022 Jul 28.
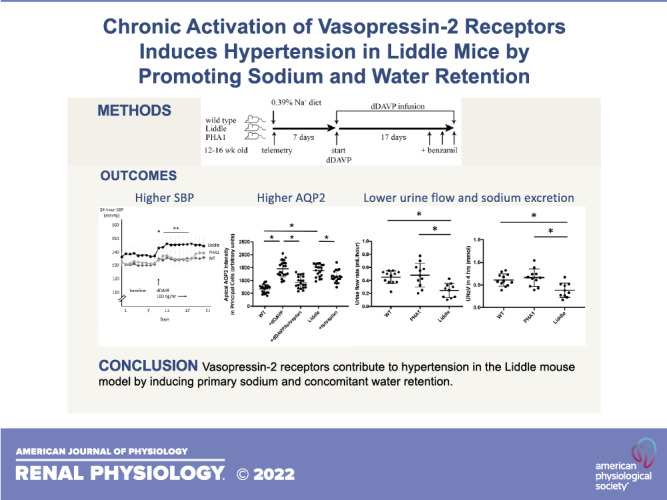
The renin-angiotensin-aldosterone and arginine vasopressin-V2 receptor-aquaporin-2 (AQP2) systems converge on the epithelial Na channel (ENaC) to regulate blood pressure and plasma tonicity. Although it is established that V2 receptors initiate renal water reabsorption through AQP2, whether V2 receptors can also induce renal Na retention through ENaC and raise blood pressure remains an open question. We hypothesized that a specific increase in V2 receptor-mediated ENaC activity can lead to high blood pressure. Our approach was to test effects of chronic activation of V2 receptors in Liddle mice, a genetic mouse model of high ENaC activity, and compare differences in ENaC activity, urine Na excretion, and blood pressure with control mice. We found that ENaC activity was elevated in Liddle mice and could not be stimulated further by administration of desmopressin (dDAVP), a V2 receptor-specific agonist. In contrast, Liddle mice showed higher levels of expression of AQP2 and aquaporin-3, but they could still respond to dDAVP infusion by increasing phospho-AQP2 expression. With dDAVP infusion, Liddle mice excreted smaller urine volume and less urine Na and developed higher blood pressure compared with control mice; this hypertension was attenuated with administration of the ENaC inhibitor benzamil. We conclude that V2 receptors contribute to hypertension in the Liddle mouse model by promoting primary Na and concomitant water retention. Liddle syndrome is a classic model for hypertension from high epithelial Na channel (ENaC) activity. In the Liddle mouse model, vasopressin-2 receptors stimulate both ENaC and aquaporin-2, which increases Na and water retention to such an extent that hypertension ensues. Liddle mice will preserve plasma tonicity at the expense of a higher blood pressure; these data highlight the inherent limitation in which the kidney must use ENaC as a pathway to regulate both plasma tonicity and blood pressure.
肾素-血管紧张素-醛固酮和精氨酸加压素-V2 受体-水通道蛋白-2(AQP2)系统集中在上皮钠通道(ENaC)上,以调节血压和血浆渗透压。虽然已经确定 V2 受体通过 AQP2 启动肾脏水重吸收,但 V2 受体是否也可以通过 ENaC 诱导肾脏钠潴留并升高血压仍然是一个悬而未决的问题。我们假设 V2 受体介导的 ENaC 活性的特定增加会导致高血压。我们的方法是在 Liddle 小鼠中测试 V2 受体的慢性激活的作用,Liddle 小鼠是一种 ENaC 活性高的遗传小鼠模型,并比较与对照小鼠相比,ENaC 活性、尿钠排泄和血压的差异。我们发现,Liddle 小鼠的 ENaC 活性升高,并且不能通过给予 V2 受体特异性激动剂去氨加压素(dDAVP)进一步刺激。相比之下,Liddle 小鼠表现出更高水平的 AQP2 和水通道蛋白-3 的表达,但它们仍然可以通过增加磷酸化 AQP2 的表达来响应 dDAVP 输注。与对照小鼠相比,给予 dDAVP 输注后,Liddle 小鼠的尿量减少,尿钠排泄减少,血压升高;这种高血压在给予 ENaC 抑制剂苯唑咪后减轻。我们的结论是,V2 受体通过促进原发性钠和伴随的水潴留来促进 Liddle 小鼠模型中的高血压。Liddle 综合征是一种经典的高上皮钠通道(ENaC)活性高血压模型。在 Liddle 小鼠模型中,加压素-2 受体刺激 ENaC 和水通道蛋白-2,从而增加钠和水的潴留,以至于高血压随之而来。Liddle 小鼠将以更高的血压为代价保持血浆渗透压;这些数据突出了肾脏必须将 ENaC 用作调节血浆渗透压和血压的途径的固有局限性。