Department of Neurosurgery, Birmingham Children's Hospital, Steelhouse Lane, Birmingham, B4 6NH, UK.
Department of Neurology, Birmingham Children's Hospital, Steelhouse Lane, Birmingham, B4 6NH, UK.
Childs Nerv Syst. 2022 Oct;38(10):1855-1859. doi: 10.1007/s00381-022-05617-1. Epub 2022 Jul 30.
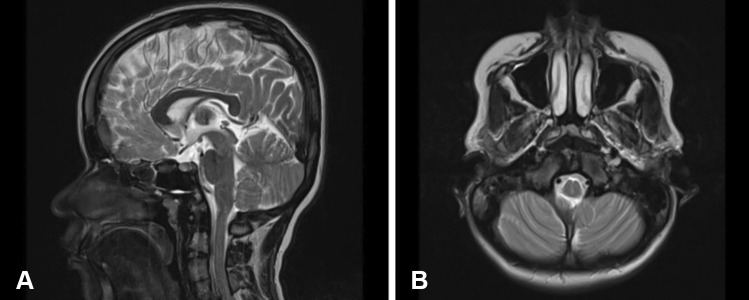
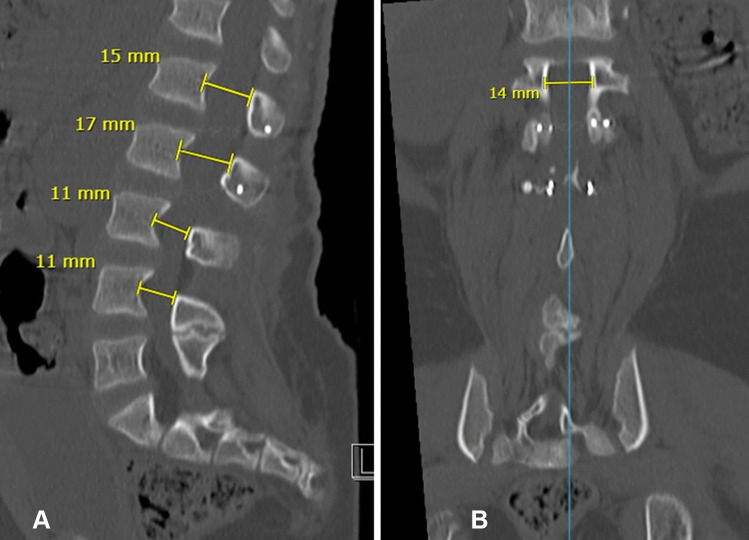
Achondroplasia is the commonest skeletal dysplasia of autosomal dominant inheritance caused by "gain of function" mutations in the fibroblast growth factor receptor 3 (FGFR3) gene. Foramen magnum compression due to accelerated ossification and spinal canal stenosis secondary to reduced interpedicular distance is a hallmark of achondroplasia, driven by G380R nucleotide pair substitution. In severe cases, limb weakness and neurogenic claudication will require surgical decompression. Rarely, a neurological condition may mimic the compressive spinal dysfunction and therefore, non-surgical causes must also be considered in cases of acute neurological deterioration in children with achondroplasia. Myasthenia gravis (MG) is an autoimmune condition resulting in fatigable muscle weakness. There are no reported cases of myasthenia gravis in achondroplasia in the literature.
We report a child with achondroplasia scheduled for decompressive surgery for severe lumbar canal stenosis presenting with neurological claudication and knee weakness. While waiting for surgery during the COVID-19 pandemic, she developed generalized fatigability and severe weakness raising concerns of acute worsening of cord compression. Urgent investigations ruled out spinal cord compression but identified an unexpected concurrent myasthenia gravis with positive antibodies to acetylcholine receptors. The surgical intervention was postponed averting the potential risk of life-threatening anaesthetic complications. She was successfully managed with a combination of pyridostigmine, steroids, azathioprine, and plasma exchange.
We report the first case of myasthenia gravis in achondroplasia and review implications in the management.
软骨发育不全症是最常见的常染色体显性遗传骨骼发育不良,由成纤维细胞生长因子受体 3 (FGFR3) 基因的“功能获得”突变引起。由于矢状窦狭窄和椎管狭窄导致的枕骨大孔压迫是软骨发育不全症的标志,这是由 G380R 核苷酸对取代驱动的。在严重的情况下,四肢无力和神经性跛行需要手术减压。在儿童中,由于神经源性跛行导致的急性神经功能恶化很少见,因此,在患有软骨发育不全症的儿童中出现急性神经恶化时,也必须考虑非手术原因。重症肌无力 (MG) 是一种自身免疫性疾病,导致易疲劳性肌肉无力。在文献中没有报道软骨发育不全症合并重症肌无力的病例。
我们报告了一例患有软骨发育不全症的儿童,该儿童因严重的腰椎管狭窄症需要进行减压手术,表现为神经源性跛行和膝关节无力。在 COVID-19 大流行期间等待手术期间,她出现了全身易疲劳和严重无力,引起了对脊髓压迫急性恶化的担忧。紧急检查排除了脊髓压迫,但发现了意外的同时存在的重症肌无力,乙酰胆碱受体抗体阳性。手术干预被推迟,避免了潜在的危及生命的麻醉并发症风险。她成功地接受了吡啶斯的明、类固醇、硫唑嘌呤和血浆置换的联合治疗。
我们报告了首例软骨发育不全症合并重症肌无力病例,并回顾了其在治疗中的意义。