Division of Nephrology and Hypertension, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Division of Nephrology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Int J Mol Sci. 2022 Jul 27;23(15):8284. doi: 10.3390/ijms23158284.
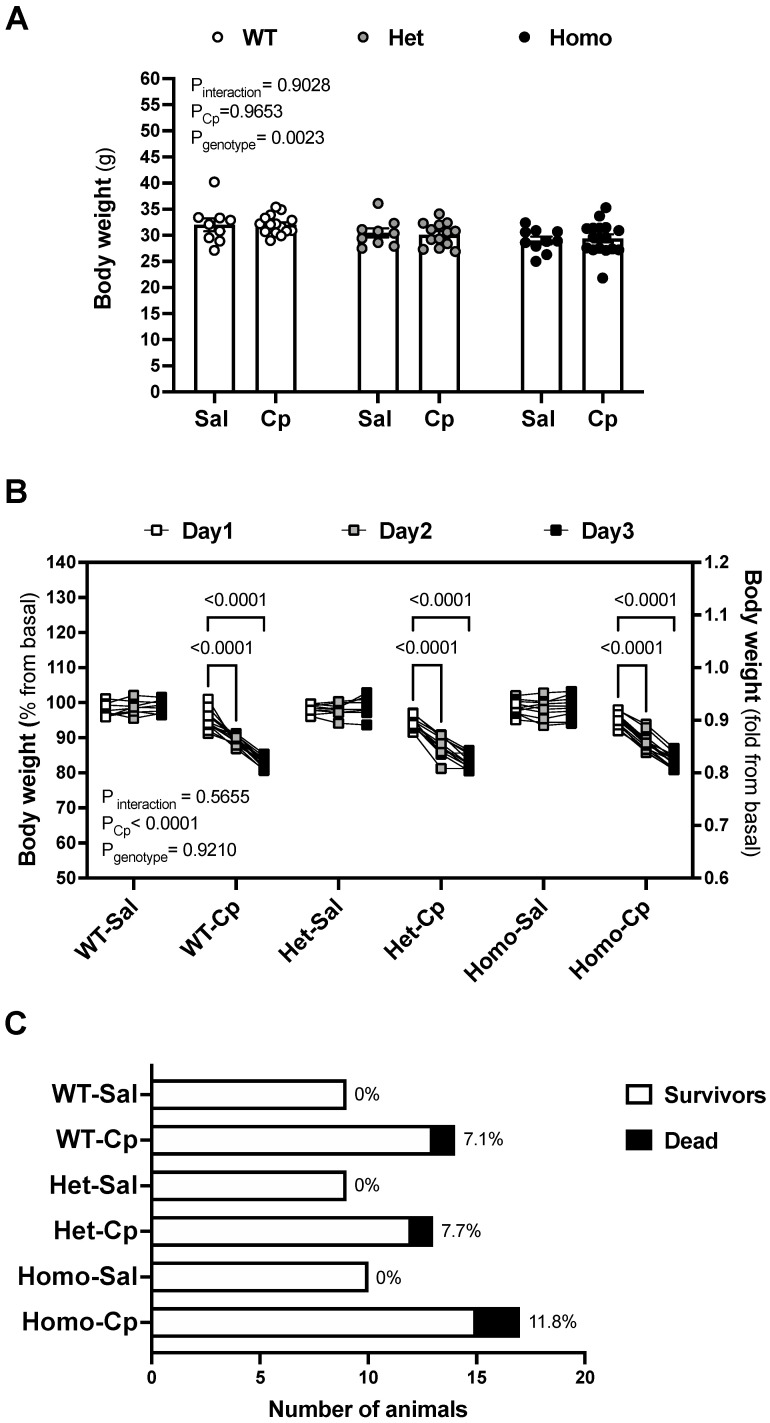
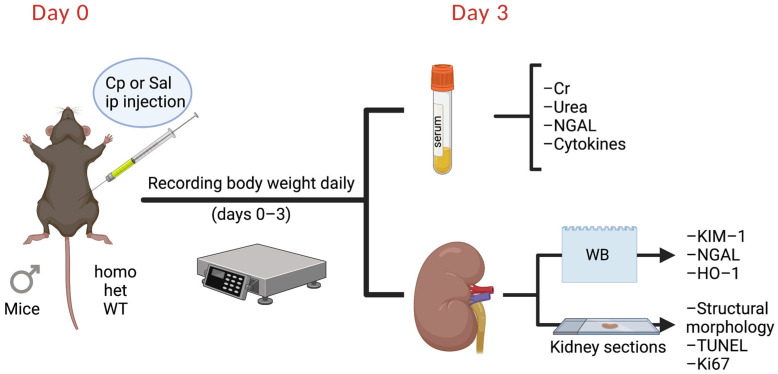
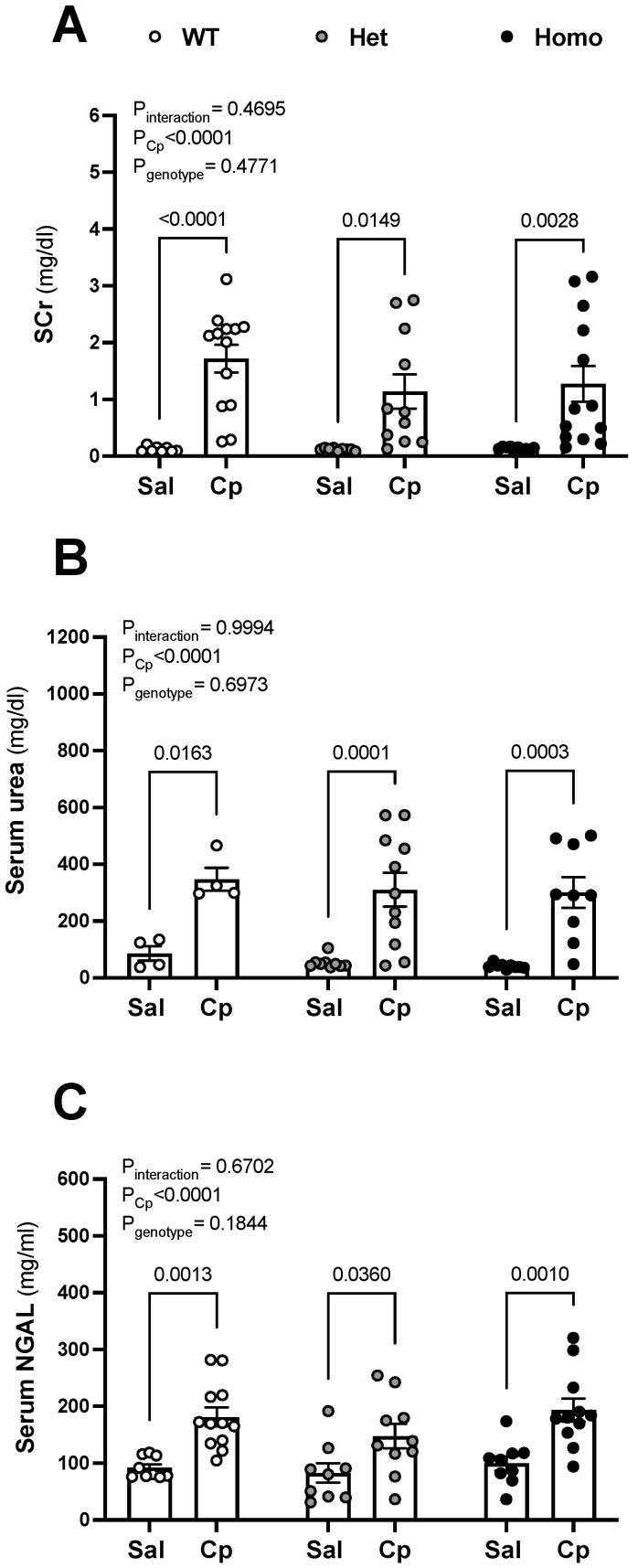
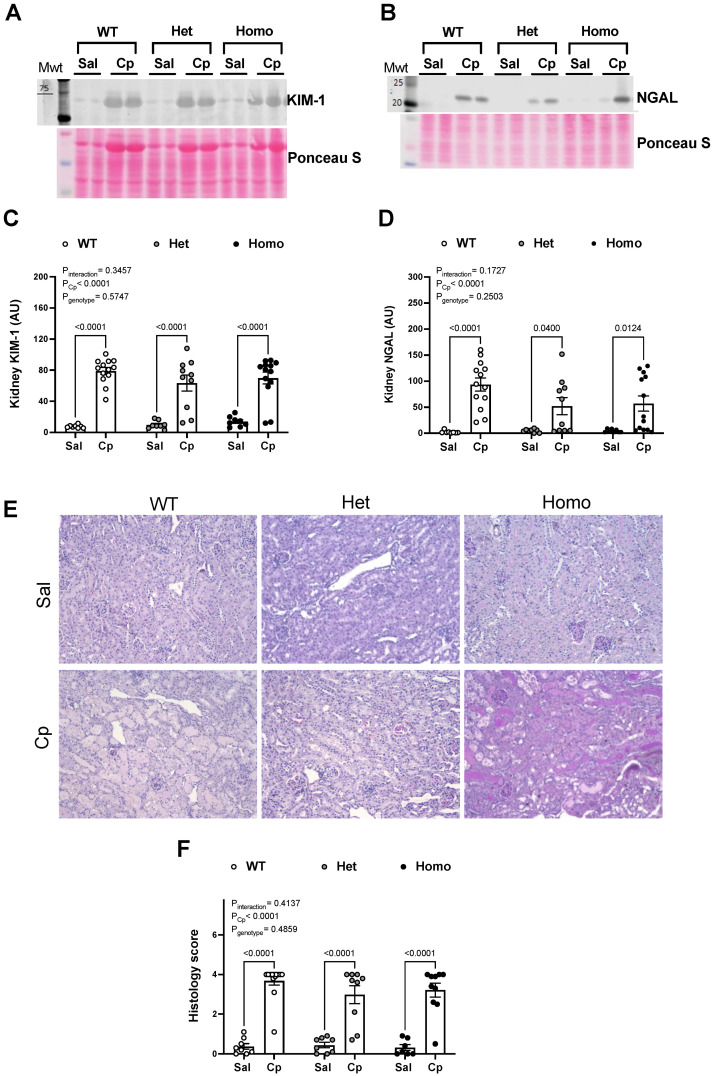
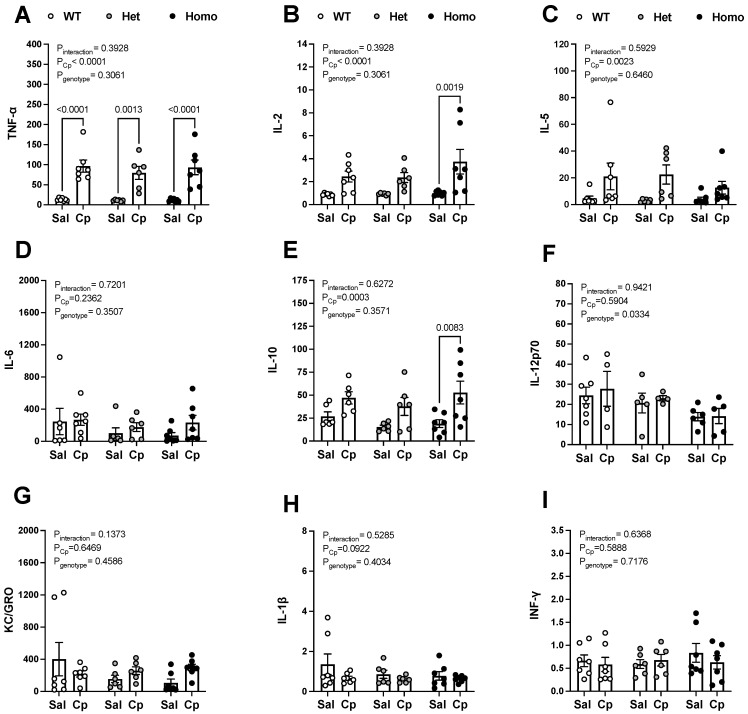
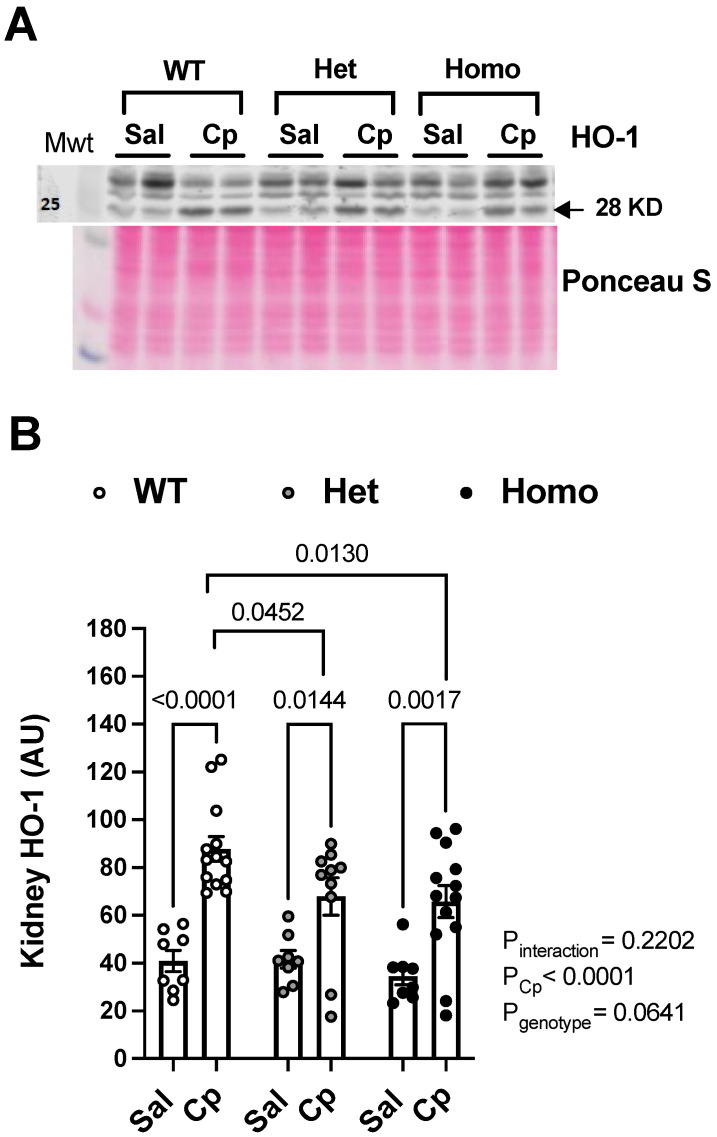
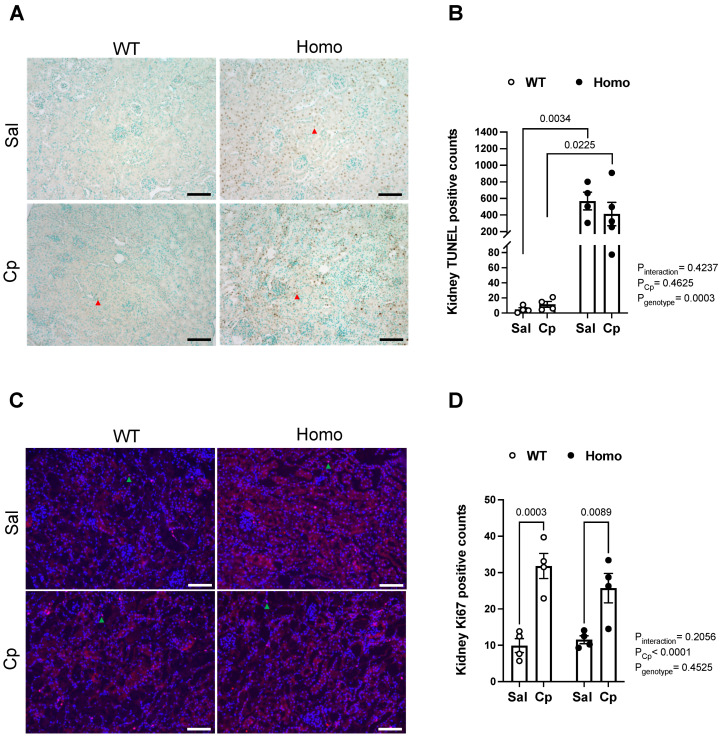
Nephrotoxicity is the dose-limiting side-effect of the chemotherapeutic agent cisplatin (Cp). Recent evidence points to renal protective actions of G protein-coupled estrogen receptor 1 (GPER1). In addition, it has been shown that GPER1 signaling elicits protective actions against acute ischemic injuries that involve multiple organ systems; however, the involvement of GPER1 signaling in Cp-induced acute kidney injury (AKI) remains unclear. This study tested whether genetic deletion of GPER1 exacerbates Cp-induced AKI in male mice. We subjected male mice, homozygous (homo) and heterozygous (het) knockout for the GPER1 gene, and wild-type (WT) littermates to Cp or saline injections and assessed markers for renal injury on the third day after injections. We also determined serum levels of proinflammatory markers in saline and Cp-treated mice. Given the protective role of heme oxygenase-1 (HO-1) in Cp-mediated apoptosis, we also investigated genotypic differences in renal HO-1 abundance, cell death, and proliferation by Western blotting, the TUNEL assay, and Ki67 immunostaining, respectively. Cp increased serum creatinine, urea, and neutrophil gelatinase-associated lipocalin (NGAL) levels, the renal abundance of kidney injury molecule-1, and NGAL in all groups. Cp-induced AKI resulted in comparable histological evidence of injury in all genotypes. WT and homo mice showed greater renal HO-1 abundance in response to Cp. Renal HO-1 abundance was lower in Cp-treated homo, compared to Cp-treated WT mice. Of note, GPER1 deletion elicited a remarkable increase in renal apoptosis; however, no genotypic differences in cell proliferation were observed. Cp augmented kidney Ki67-positive counts, regardless of the genotype. Overall, our data do not support a role for GPER1 in mediating Cp-induced renal injury. GPER1 deletion promotes renal apoptosis and diminishes HO-1 induction in response to Cp, suggesting that GPER1 may play cytoprotective and anti-apoptotic actions in AKI. GPER1-induced regulation of HO-1 and apoptosis may offer novel therapeutic targets for the treatment of AKI.
肾毒性是化疗药物顺铂(Cp)的剂量限制副作用。最近的证据表明 G 蛋白偶联雌激素受体 1(GPER1)具有肾脏保护作用。此外,已经表明 GPER1 信号转导可以针对涉及多个器官系统的急性缺血性损伤产生保护作用;然而,GPER1 信号转导在 Cp 诱导的急性肾损伤(AKI)中的参与仍不清楚。本研究测试了 GPER1 基因缺失是否会加重雄性小鼠的 Cp 诱导的 AKI。我们使雄性小鼠、GPER1 基因纯合(homo)和杂合(het)敲除小鼠以及野生型(WT)同窝小鼠接受 Cp 或生理盐水注射,并在注射后第 3 天评估肾脏损伤标志物。我们还测定了生理盐水和 Cp 处理小鼠血清中促炎标志物的水平。鉴于血红素加氧酶-1(HO-1)在 Cp 介导的细胞凋亡中的保护作用,我们还通过 Western 印迹、TUNEL 测定和 Ki67 免疫染色分别研究了肾脏 HO-1 丰度、细胞死亡和增殖的基因型差异。Cp 增加了血清肌酐、尿素和中性粒细胞明胶酶相关脂质运载蛋白(NGAL)水平,所有组的肾脏损伤分子-1 和 NGAL 的肾脏丰度。Cp 诱导的 AKI 在所有基因型中均导致相似的损伤组织学证据。WT 和 homo 小鼠对 Cp 的反应显示出更高的肾脏 HO-1 丰度。与 Cp 处理的 WT 小鼠相比,Cp 处理的 homo 小鼠的肾脏 HO-1 丰度更低。值得注意的是,GPER1 缺失引起明显的肾脏细胞凋亡增加;然而,细胞增殖没有基因型差异。Cp 增加了肾脏 Ki67 阳性计数,与基因型无关。总的来说,我们的数据不支持 GPER1 在介导 Cp 诱导的肾脏损伤中的作用。GPER1 缺失促进了 Cp 诱导的肾脏细胞凋亡,并减少了对 Cp 的 HO-1 诱导,这表明 GPER1 可能在 AKI 中发挥细胞保护和抗细胞凋亡作用。GPER1 诱导的 HO-1 和细胞凋亡的调节可能为 AKI 的治疗提供新的治疗靶点。