Orthopedic Research Institute, Department of Orthopedics, Sichuan University West China Hospital, Chengdu, China.
Front Immunol. 2022 Jul 28;13:941398. doi: 10.3389/fimmu.2022.941398. eCollection 2022.
Juvenile idiopathic arthritis (JIA) is the most common rheumatic disease in children, and its pathogenesis is still unclear. Genome-wide association studies (GWASs) of JIA have identified hundreds of risk factors, but few of them implicated specific biological mechanisms.
A cross-tissue transcriptome-wide association study (TWAS) was performed with the functional summary-based imputation software (FUSION) tool based on GWAS summary datasets (898 JIA patients and 346,102 controls from BioBank Japan (BBJ)/FinnGen). The gene expression reference weights of skeletal muscle and the whole blood were obtained from the Genotype-Tissue Expression (GTExv8) project. JIA-related genes identified by TWAS findings genes were further compared with the differentially expressed genes (DEGs) identified by the mRNA expression profile of JIA from the Gene Expression Omnibus (GEO) database (accession number: GSE1402). Last, candidate genes were analyzed using functional enrichment and annotation analysis by Metascape to examine JIA-related gene sets.
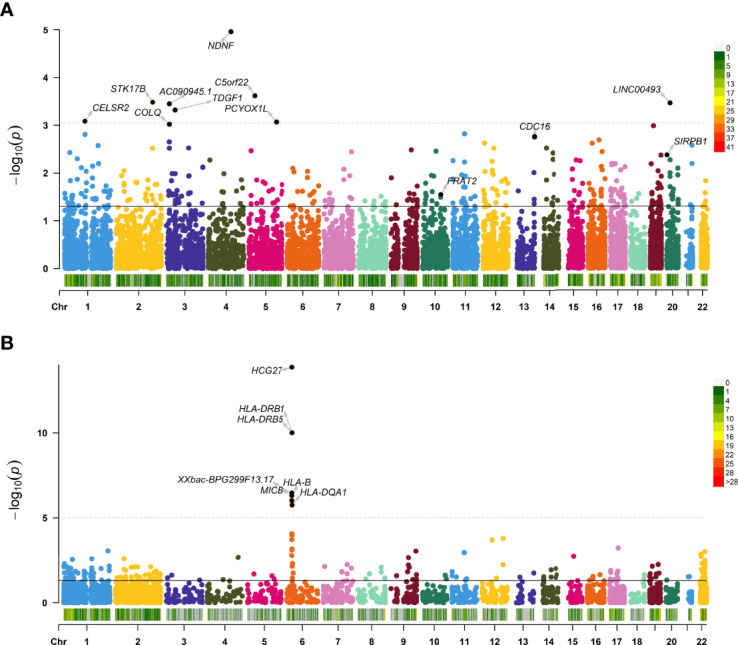
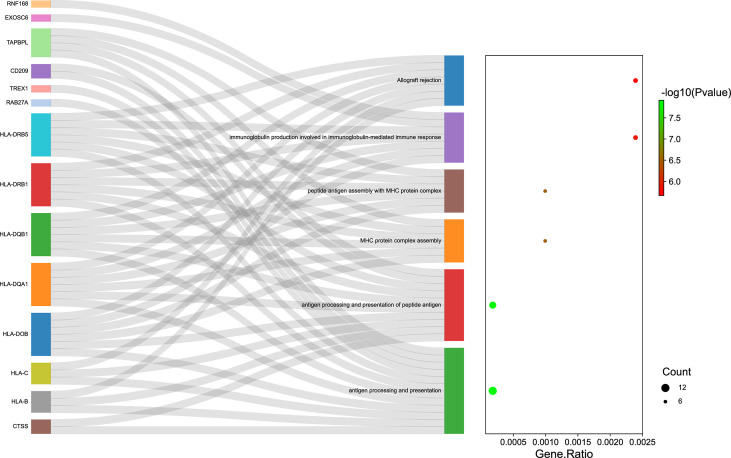
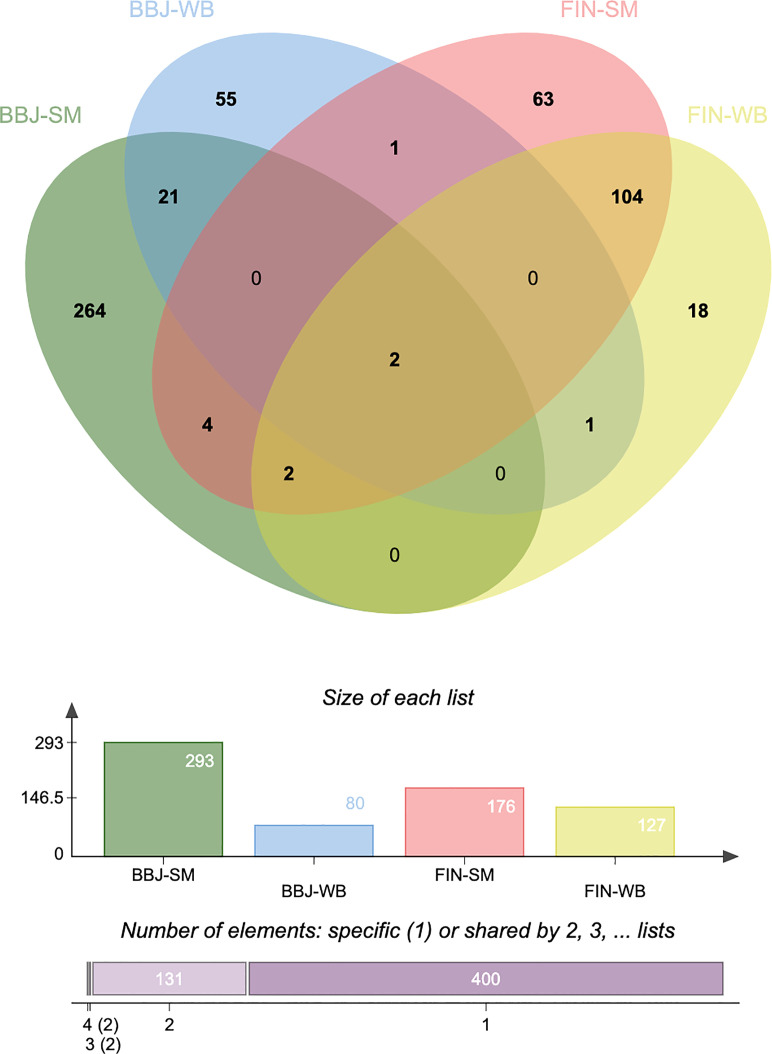
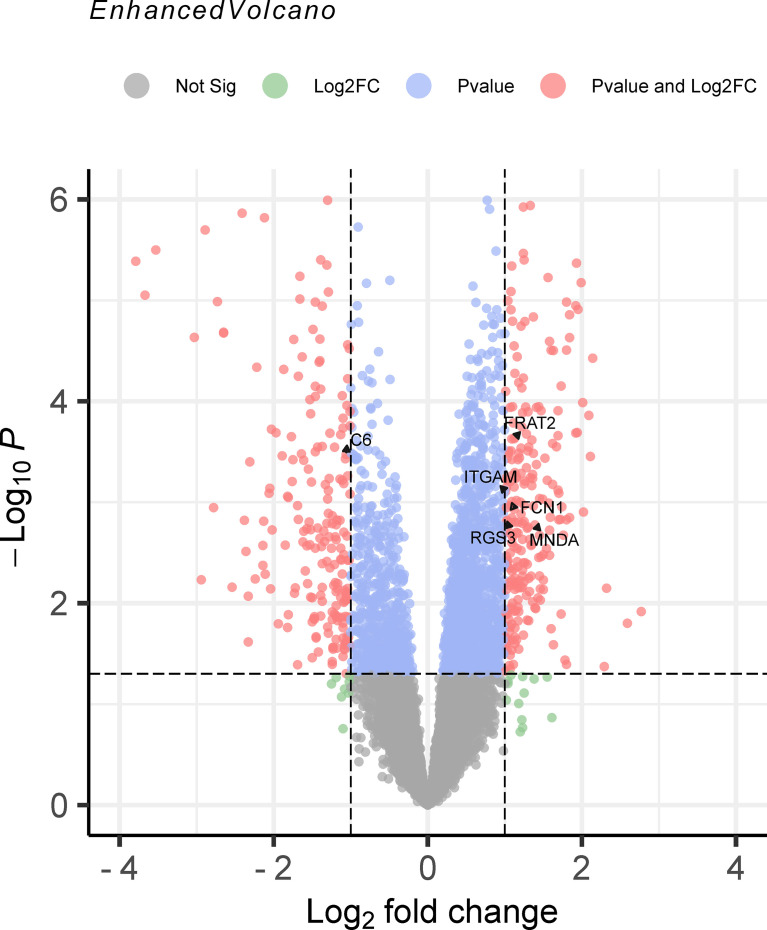
The TWAS identified 535 significant genes with < 0.05 and contains 350 for Asian and 195 for European (including 10 genes both expressed in Asian and European), such as ( = 1.72E-03) and ( = 3.65E-02). Eight overlapping genes were identified based on TWAS results and DEGs of JIA patients, such as ( = 4.21E-03, = 1.50E-04) and ( = 2.82E-02, = 1.43E-02). Pathway enrichment analysis of TWAS identified 183 pathways such as cytokine signaling in the immune system and cell adhesion molecules. By integrating the results of DEGs pathway and process enrichment analyses, 19 terms were identified such as positive regulation of T-cell activation.
By conducting two populations TWAS, we identified a group of JIA-associated genes and pathways, which may provide novel clues to uncover the pathogenesis of JIA.
幼年特发性关节炎(JIA)是儿童中最常见的风湿性疾病,其发病机制尚不清楚。JIA 的全基因组关联研究(GWAS)已经确定了数百个风险因素,但其中很少有涉及特定的生物学机制。
使用基于全基因组关联研究汇总数据集(来自日本生物银行/芬兰遗传(BBJ/FinnGen)的 898 名 JIA 患者和 346102 名对照)的功能汇总基于的跨组织转录组全关联研究(TWAS)进行分析(TWAS)。骨骼肌和全血的基因表达参考权重来自基因型组织表达(GTExv8)项目。TWAS 发现的与 JIA 相关的基因与从基因表达综合数据库(GEO)(注册号:GSE1402)获得的 JIA 的 mRNA 表达谱中确定的差异表达基因(DEGs)进一步比较。最后,使用 Metascape 进行功能富集和注释分析来分析候选基因,以检查与 JIA 相关的基因集。
TWAS 确定了 535 个具有 <0.05 的显著基因,其中 350 个基因在亚洲人群中表达,195 个基因在欧洲人群中表达(包括 10 个在亚洲和欧洲人群中均表达的基因),例如 ( = 1.72E-03)和 ( = 3.65E-02)。根据 TWAS 结果和 JIA 患者的 DEGs 确定了 8 个重叠基因,例如 ( = 4.21E-03, = 1.50E-04)和 ( = 2.82E-02, = 1.43E-02)。TWAS 的通路富集分析鉴定了 183 条通路,如免疫系统中的细胞因子信号传导和细胞黏附分子。通过整合 DEGs 通路和过程富集分析的结果,确定了 19 个术语,如 T 细胞激活的正调控。
通过对两个人群进行 TWAS,我们确定了一组与 JIA 相关的基因和通路,这可能为揭示 JIA 的发病机制提供新的线索。