Frizinsky Shirly, Rechavi Erez, Barel Ortal, Lee Yu Nee, Simon Amos J, Lev Atar, Stauber Tali, Adam Etai, Somech Raz
Pediatric Department A and the Immunology Service, Jeffrey Modell Foundation Center, Edmond and Lily Safra Children's Hospital, Sheba Medical Center, Ramat Gan, Israel.
Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel.
Front Pediatr. 2022 Jul 27;10:883173. doi: 10.3389/fped.2022.883173. eCollection 2022.
During the process of generating diverse T and B cell receptor (TCR and BCR, respectively) repertoires, double-strand DNA breaks are produced. Subsequently, these breaks are corrected by a complex system led by the non-homologous end-joining (NHEJ). Pathogenic variants in genes involved in this process, such as the gene, cause severe combined immunodeficiency syndrome (SCID) along with neurodevelopmental disease and sensitivity to ionizing radiation.
To provide new clinical and immunological insights on NHEJ1 deficiency arising from a newly diagnosed patient with severe immunodeficiency.
A male infant, born to consanguineous parents, suspected of having primary immunodeficiency underwent immunological and genetic workup. This included a thorough assessment of T cell phenotyping and lymphocyte activation by mitogen stimulation tests, whole-exome sequencing (WES), TCR repertoire Vβ repertoire flow cytometry analysis, and TCR and BCR repertoire analysis next-generation sequencing (NGS).
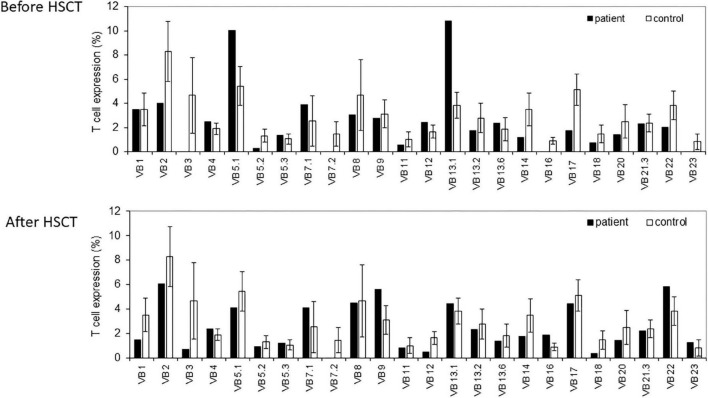
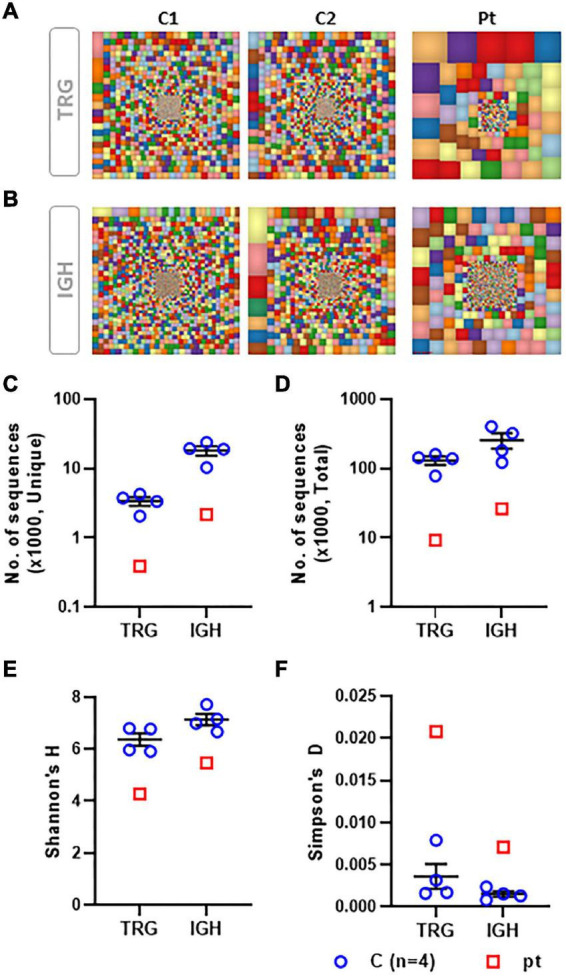
Clinical findings included microcephaly, recurrent pneumonia, and failure to thrive. An immune workup revealed lymphopenia, reduced T cell function, and hypogammaglobulinemia. Skewed TCR Vβ repertoire, TCR gamma (TRG) repertoire, and BCR repertoire were determined in the patient. Genetic analysis identified a novel homozygous missense pathogenic variant in : c.A580Ins.T; p.M194fs. The patient underwent a successful hematopoietic stem cell transplantation (HSCT).
A novel pathogenic variant is reported in a patient who presented with SCID phenotype that displayed clonally expanded T and B cells. An adjusted HSCT was safe to ensure full T cell immune reconstitution.
在产生多样化的T细胞和B细胞受体(分别为TCR和BCR)库的过程中,会产生双链DNA断裂。随后,这些断裂由非同源末端连接(NHEJ)主导的复杂系统进行修复。参与此过程的基因中的致病变异,如该基因,会导致严重联合免疫缺陷综合征(SCID)以及神经发育疾病和对电离辐射的敏感性。
为一名新诊断的严重免疫缺陷患者出现的NHEJ1缺乏症提供新的临床和免疫学见解。
一名父母近亲结婚的男婴,怀疑患有原发性免疫缺陷,接受了免疫学和遗传学检查。这包括通过丝裂原刺激试验对T细胞表型和淋巴细胞活化进行全面评估、全外显子测序(WES)、TCR库Vβ库流式细胞术分析以及TCR和BCR库分析下一代测序(NGS)。
临床发现包括小头畸形、反复肺炎和发育不良。免疫检查显示淋巴细胞减少、T细胞功能降低和低丙种球蛋白血症。患者的TCR Vβ库、TCRγ(TRG)库和BCR库存在偏态分布。基因分析在该基因中鉴定出一种新的纯合错义致病变异:c.A580Ins.T;p.M194fs。患者接受了成功的造血干细胞移植(HSCT)。
在一名表现出SCID表型且显示T和B细胞克隆性扩增的患者中报告了一种新的致病变异。调整后的HSCT对于确保完全的T细胞免疫重建是安全的。