Musa Klefah A K, Eriksson Leif A
Department of Medicinal Chemistry, Pharmacy College, El-Mergib University, Al-Khoms 18342, Libya.
Department of Chemistry and Molecular Biology, University of Gothenburg, 405 30 Göteborg, Sweden.
ACS Omega. 2022 Aug 11;7(33):29475-29482. doi: 10.1021/acsomega.2c03118. eCollection 2022 Aug 23.
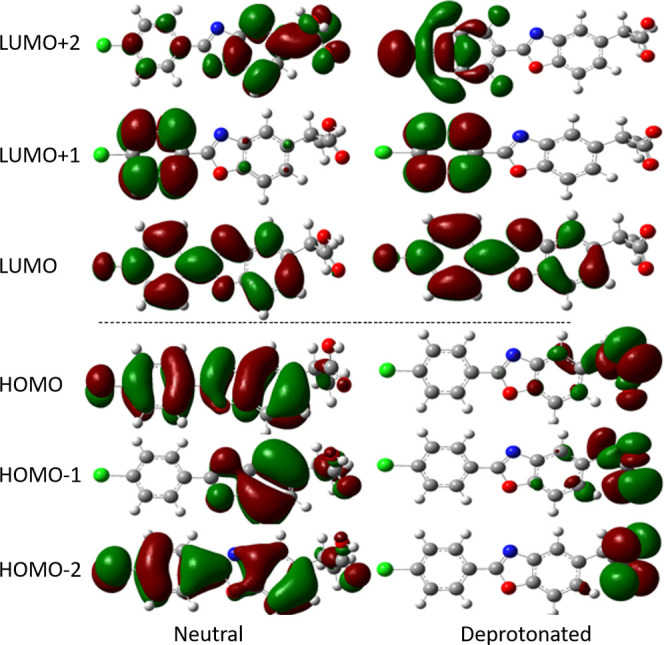
Computational quantum chemistry within the density functional theory (DFT) and time-dependent density functional theory (TD-DFT) framework is used to investigate the photodegradation mechanism as well as the photochemical and photophysical properties of benoxaprofen (BP), a non steroid anti-inflammatory molecule (2-[2-(4-chlorophenyl)-1,3-benzoxazol-5-yl] propanoic acid). BP is a highly phototoxic agent that causes cutaneous phototoxicity shortly after its administration. On the grounds of concern about serious side effects, especially hepatotoxicity, it was withdrawn from the world market after only 2 years of its release. Our study shows that the drug has the capability to absorb radiation in the UV region, mainly between 300 and 340 nm, and undergoes spontaneous photoinduced decarboxylation from the triplet state. It shows very similar photochemical properties to the highly photolabile non-steroidal anti-inflammatory drugs (NSAIDs) ketoprofen, suprofen, and tiaprofenic acid. Like ketoprofen, BP can also decarboxylate from excited singlet states by overcoming low energy barriers. The differences in molecular orbital (highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO)) distributions between the neutral and deprotonated BP, their absorption spectra, and the energetics and fate of various photoproducts produced throughout the photodegradation are discussed. Initiation and termination of decarboxylated BP radical species and initiation of propagating lipid peroxidation reactions due to the addition of molecular oxygen giving rise to the corresponding peroxyl radical are also explored in detail.
在密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)框架内的计算量子化学方法,被用于研究非甾体抗炎分子苯氧布洛芬(BP,2-[2-(4-氯苯基)-1,3-苯并恶唑-5-基]丙酸)的光降解机理以及光化学和光物理性质。BP是一种高光毒性药物,给药后不久就会引起皮肤光毒性。出于对其严重副作用尤其是肝毒性的担忧,该药物在上市仅两年后就退出了世界市场。我们的研究表明,该药物有能力吸收紫外区域的辐射,主要在300至340纳米之间,并从三重态发生自发的光诱导脱羧反应。它表现出与高光不稳定的非甾体抗炎药(NSAIDs)酮洛芬、舒洛芬和噻洛芬酸非常相似的光化学性质。与酮洛芬一样,BP也能通过克服低能垒从激发单重态脱羧。讨论了中性和去质子化BP之间分子轨道(最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO))分布的差异、它们的吸收光谱,以及光降解过程中产生的各种光产物的能量学和归宿。还详细探讨了脱羧BP自由基物种的引发和终止,以及由于分子氧的加入引发脂质过氧化反应并产生相应过氧自由基的过程。