Department of Pediatric Neurology, Vall d'Hebron Hospital Universitary and Vall d'Hebrón Research Institute (VHIR)., Barcelona, Spain.
Department of Pediatrics, Obstetrics, Gynecology, Preventative Medicine and Public Health, Universitat Autònoma de Barcelona, Barcelona, Spain.
Mov Disord. 2022 Nov;37(11):2197-2209. doi: 10.1002/mds.29182. Epub 2022 Aug 25.
The objective of this study was to better delineate the genetic landscape and key clinical characteristics of complex, early-onset, monogenic hyperkinetic movement disorders.
Patients were recruited from 14 international centers. Participating clinicians completed standardized proformas capturing demographic, clinical, and genetic data. Two pediatric movement disorder experts reviewed available video footage, classifying hyperkinetic movements according to published criteria.
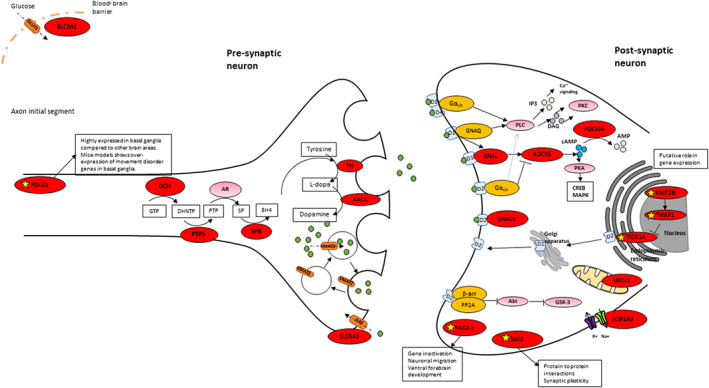
One hundred forty patients with pathogenic variants in 17 different genes (ADCY5, ATP1A3, DDC, DHPR, FOXG1, GCH1, GNAO1, KMT2B, MICU1, NKX2.1, PDE10A, PTPS, SGCE, SLC2A1, SLC6A3, SPR, and TH) were identified. In the majority, hyperkinetic movements were generalized (77%), with most patients (69%) manifesting combined motor semiologies. Parkinsonism-dystonia was characteristic of primary neurotransmitter disorders (DDC, DHPR, PTPS, SLC6A3, SPR, TH); chorea predominated in ADCY5-, ATP1A3-, FOXG1-, NKX2.1-, SLC2A1-, GNAO1-, and PDE10A-related disorders; and stereotypies were a prominent feature in FOXG1- and GNAO1-related disease. Those with generalized hyperkinetic movements had an earlier disease onset than those with focal/segmental distribution (2.5 ± 0.3 vs. 4.7 ± 0.7 years; P = 0.007). Patients with developmental delay also presented with hyperkinetic movements earlier than those with normal neurodevelopment (1.5 ± 2.9 vs. 4.7 ± 3.8 years; P < 0.001). Effective disease-specific therapies included dopaminergic agents for neurotransmitters disorders, ketogenic diet for glucose transporter deficiency, and deep brain stimulation for SGCE-, KMT2B-, and GNAO1-related hyperkinesia.
This study highlights the complex phenotypes observed in children with genetic hyperkinetic movement disorders that can lead to diagnostic difficulty. We provide a comprehensive analysis of motor semiology to guide physicians in the genetic investigation of these patients, to facilitate early diagnosis, precision medicine treatments, and genetic counseling. © 2022 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
本研究旨在更详细地描绘复杂、早发性、单基因遗传性运动障碍的遗传图谱和关键临床特征。
从 14 个国际中心招募患者。参与的临床医生完成了标准化的表格,记录了人口统计学、临床和遗传数据。两名儿科运动障碍专家查看了可用的视频片段,根据已发表的标准对多动进行分类。
在 17 个不同基因(ADCY5、ATP1A3、DDC、DHPR、FOXG1、GCH1、GNAO1、KMT2B、MICU1、NKX2.1、PDE10A、PTPS、SGCE、SLC2A1、SLC6A3、SPR 和 TH)中发现了 140 名携带致病性变异的患者。在大多数患者中,多动表现为全身性(77%),大多数患者(69%)表现为运动半侧化。帕金森病-肌张力障碍是原发性神经递质紊乱的特征(DDC、DHPR、PTPS、SLC6A3、SPR、TH);舞蹈病在 ADCY5、ATP1A3、FOXG1、NKX2.1、SLC2A1、GNAO1 和 PDE10A 相关疾病中更为突出;刻板行为是 FOXG1 和 GNAO1 相关疾病的一个突出特征。全身性多动患者的发病年龄早于局灶性/节段性分布患者(2.5±0.3 岁比 4.7±0.7 岁;P=0.007)。伴有发育迟缓的患者的多动发病年龄也早于神经发育正常的患者(1.5±2.9 岁比 4.7±3.8 岁;P<0.001)。有效的疾病特异性治疗方法包括神经递质紊乱的多巴胺能药物、葡萄糖转运体缺乏的生酮饮食和 SGCE、KMT2B 和 GNAO1 相关多动的深部脑刺激。
本研究强调了儿童遗传性运动障碍的复杂表型,这可能导致诊断困难。我们对运动半侧化进行了全面分析,以指导医生对这些患者进行基因调查,从而促进早期诊断、精准医学治疗和遗传咨询。