Department of Endocrinology, Endocrinology Research Center, Xiangya Hospital, Central South University, Changsha, Hunan, China.
Department of Endocrinology, Endocrinology Research Center, Xiangya Hospital, Central South University, Changsha, Hunan, China; National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Changsha, Hunan, China.
Cell Mol Gastroenterol Hepatol. 2022;14(6):1213-1233. doi: 10.1016/j.jcmgh.2022.08.011. Epub 2022 Sep 2.
BACKGROUND & AIMS: Nonalcoholic fatty liver disease (NAFLD) is a major cause of liver-related morbidity and mortality whereas the pathogenic mechanism remains largely elusive. DNA N6-methyladenosine (6mA) modification is a recently identified epigenetic mark indicative of transcription in eukaryotic genomes. Here, we aimed to investigate the role and mechanism of DNA 6mA modification in NAFLD progression.
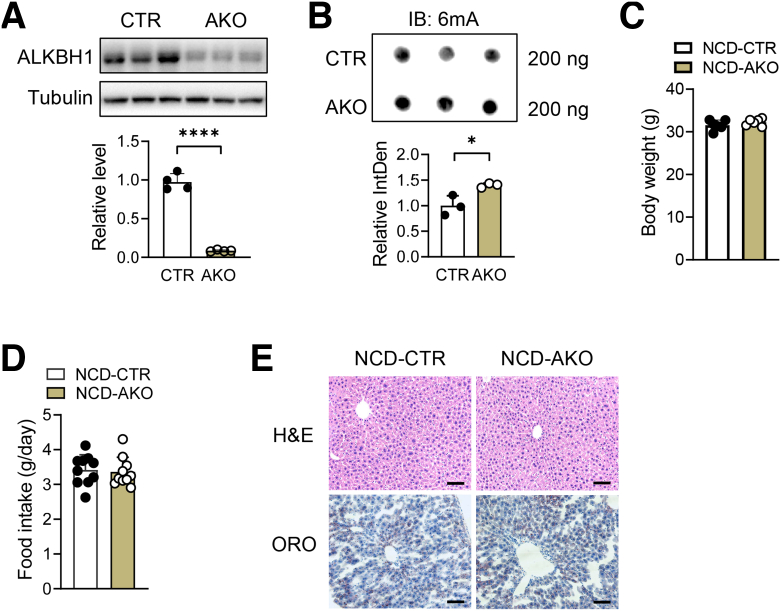
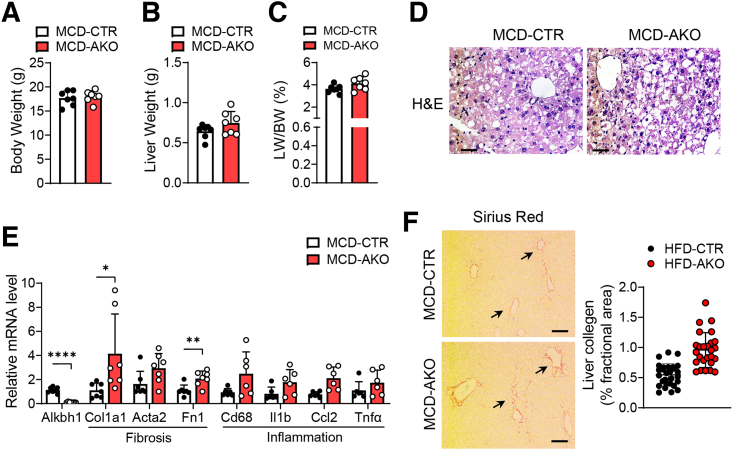
Dot blot and immunohistochemistry were used to detect DNA 6mA levels. Liver-specific AlkB homolog 1 (ALKBH1)-knockout mice and mice with ALKBH1 overexpression in liver were subjected to a high-fat diet or methionine choline-deficient diet to evaluate the critical role of ALKBH1-demethylated DNA 6mA modification in the pathogenesis of hepatic steatosis during NAFLD. RNA sequencing and chromatin immunoprecipitation sequencing were performed to investigate molecular mechanisms underlying this process.
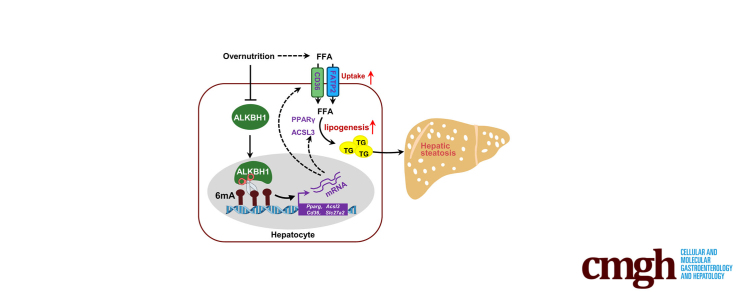
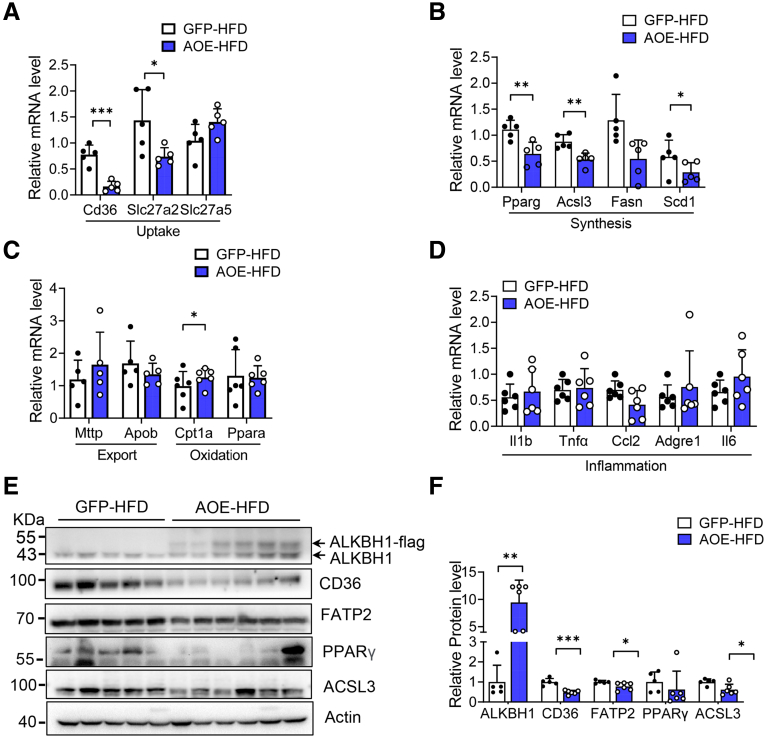
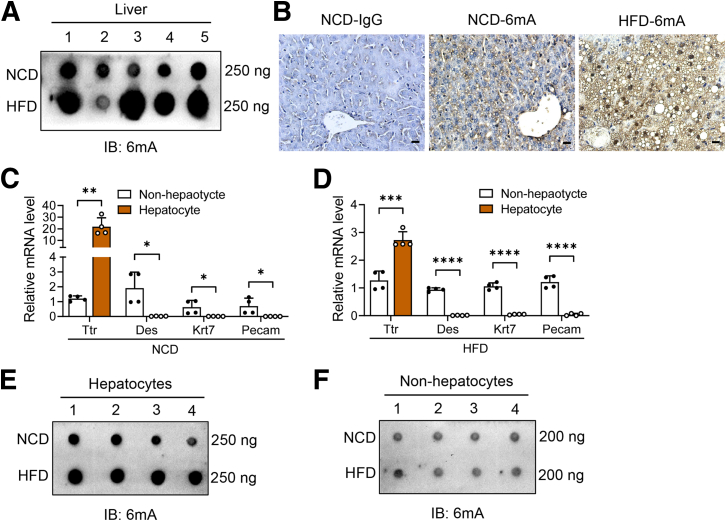
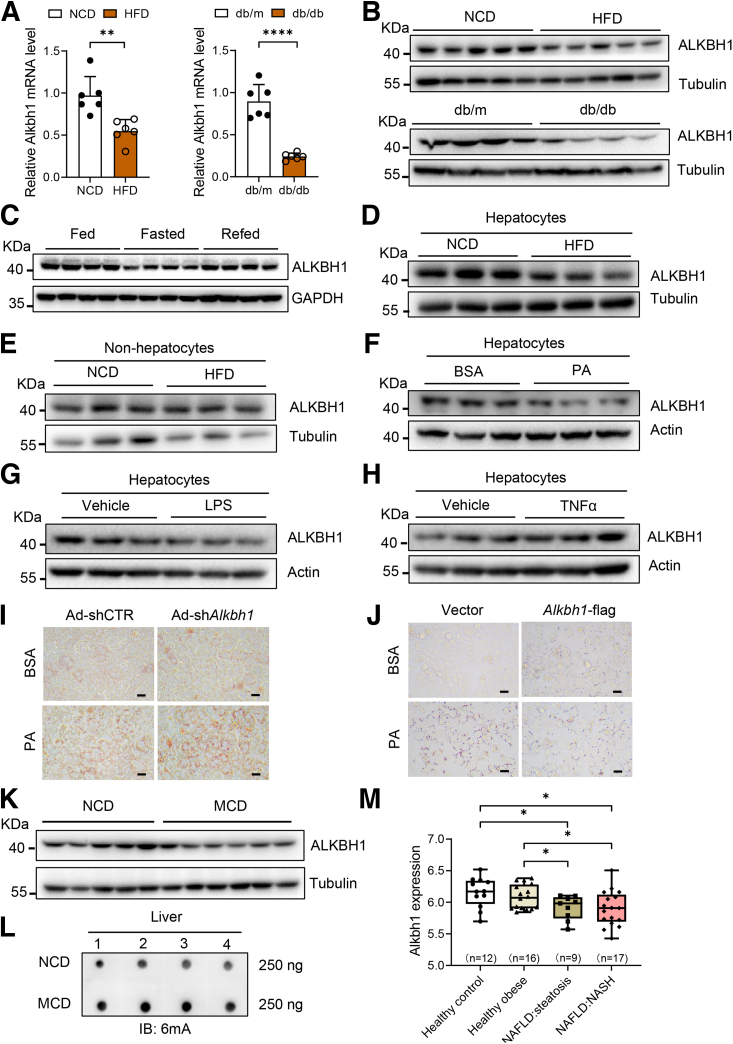
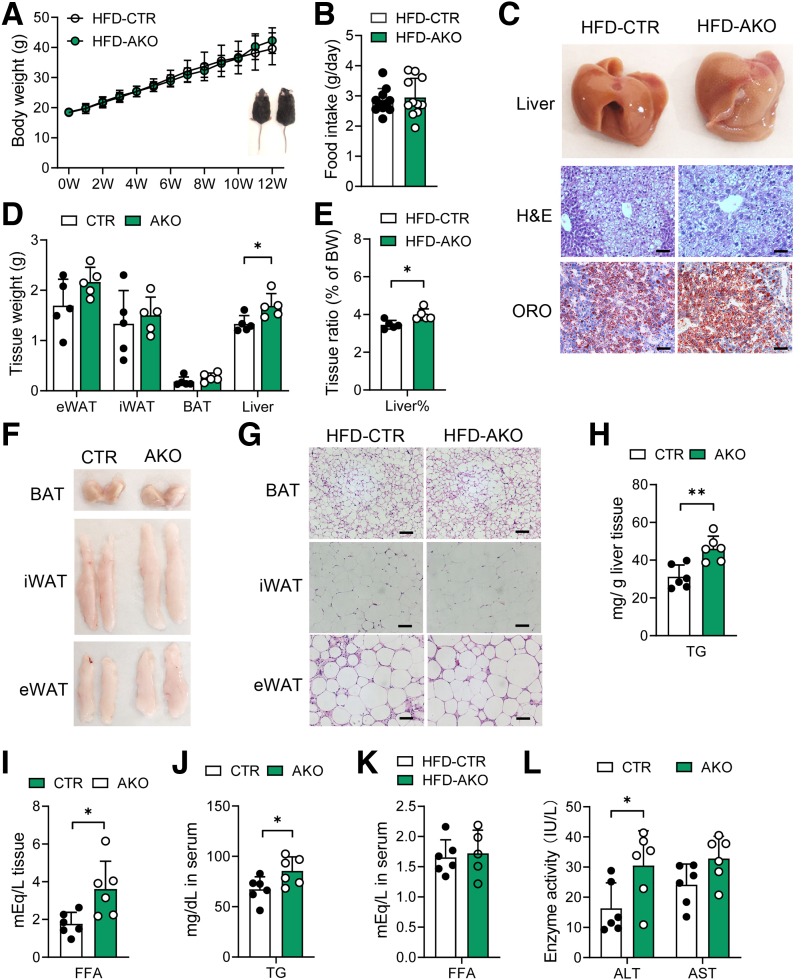
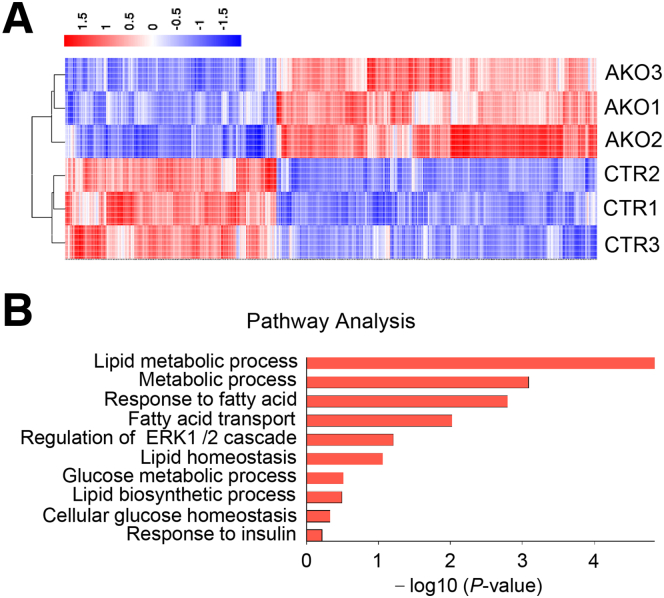
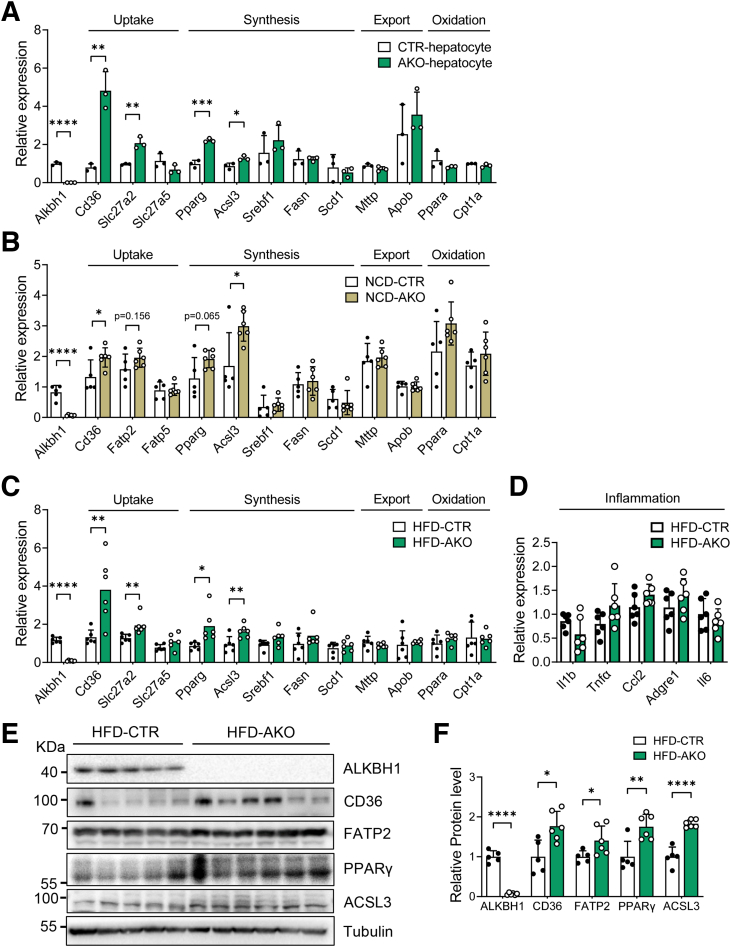
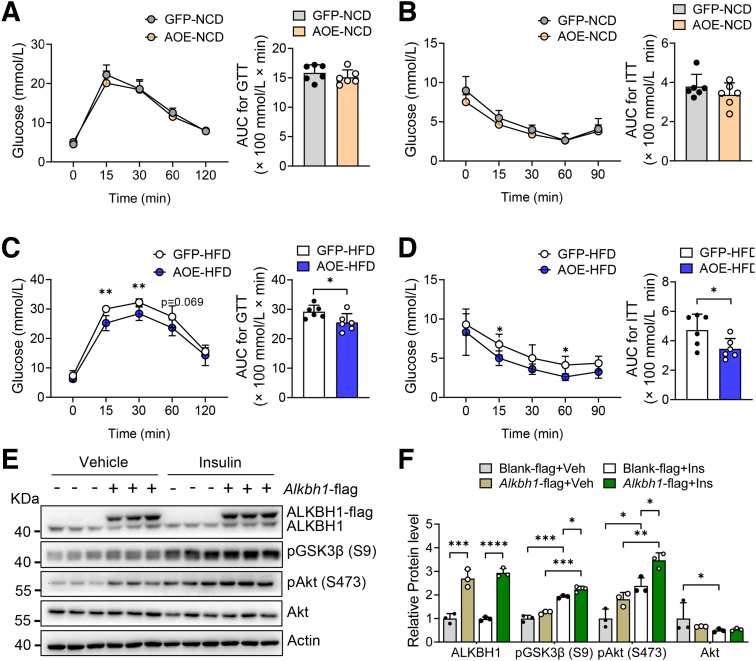
The DNA 6mA level was increased significantly with hepatic steatosis, while ALKBH1 expression was down-regulated markedly in both mouse and human fatty liver. Deletion of ALKBH1 in hepatocytes increased genomic 6mA levels and accelerated diet-induced hepatic steatosis and metabolic dysfunction. Comprehensive analyses of transcriptome and chromatin immunoprecipitation sequencing data indicated that ALKBH1 directly bound to and exclusively demethylated 6mA levels of genes involved in fatty acid uptake and lipogenesis, leading to reduced hepatic lipid accumulation. Importantly, ALKBH1 overexpression was sufficient to suppress lipid uptake and synthesis, and alleviated diet-induced hepatic steatosis and insulin resistance.
Our findings show an indispensable role of ALKBH1 as an epigenetic suppressor of DNA 6mA in hepatic fatty acid metabolism and offer a potential therapeutic target for NAFLD treatment.
非酒精性脂肪性肝病(NAFLD)是导致肝相关发病率和死亡率的主要原因,但其发病机制仍很大程度上难以捉摸。DNA N6-甲基腺嘌呤(6mA)修饰是一种最近发现的真核基因组转录的表观遗传标记。在此,我们旨在研究 DNA 6mA 修饰在 NAFLD 进展中的作用和机制。
采用点印迹和免疫组织化学方法检测 DNA 6mA 水平。采用肝特异性 AlkB 同源物 1(ALKBH1)敲除小鼠和肝中过表达 ALKBH1 的小鼠进行高脂肪饮食或蛋氨酸胆碱缺乏饮食,以评估 ALKBH1 去甲基化 DNA 6mA 修饰在 NAFLD 期间肝脂肪变性发病机制中的关键作用。进行 RNA 测序和染色质免疫沉淀测序,以研究该过程的分子机制。
随着肝脂肪变性,DNA 6mA 水平显著增加,而 ALKBH1 在人和小鼠脂肪肝中的表达明显下调。肝细胞中 ALKBH1 的缺失增加了基因组 6mA 水平,并加速了饮食诱导的肝脂肪变性和代谢功能障碍。转录组和染色质免疫沉淀测序数据的综合分析表明,ALKBH1 直接结合并特异性地去甲基化参与脂肪酸摄取和脂肪生成的基因的 6mA 水平,导致肝脂质积累减少。重要的是,ALKBH1 的过表达足以抑制脂质摄取和合成,并缓解饮食诱导的肝脂肪变性和胰岛素抵抗。
我们的研究结果表明,ALKBH1 作为 DNA 6mA 的表观遗传抑制剂,在肝脏脂肪酸代谢中起着不可或缺的作用,并为 NAFLD 的治疗提供了一个潜在的治疗靶点。